- 酪胺酸血症第1、2型 疾病介紹及衛教
酪胺酸血症是一種遺傳性代謝疾病,依照突變基因不同區分型別;第1型的致病原因為第15號染色體上的FAH基因突變,造成蛋白質功能失常,無法將延胡索醯乙醯乙酸轉換成延胡索酸和乙醯乙酸
- α1-抗胰蛋白酶缺乏症 疾病介紹及衛教
α1-抗胰蛋白酶缺乏症是一種遺傳性內分泌疾病,致病原因為第14號染色體上的SERPINA1基因突變,導致製造α1-抗胰蛋白酶(α-1 antitrypsin,AAT)功能異常或低
- 線狀體肌肉病變第2型 疾病介紹及衛教
線狀體肌肉病變第2型是一種非進行性的先天性神經肌肉病變,致病原因為第2號染色體上的NEB基因變異所致,使得Nebulin蛋白製造不足或功能降低。
- 短鏈醯輔酶Α去氫酶缺乏症 疾病介紹及衛教
短鏈醯輔酶Α去氫酶缺乏症是一種脂肪酸代謝異常疾病,致病原因為第12號染色體上的ACADS基因變異所致,導致短鏈醯輔酶Α去氫酶(SCAD)缺乏或功能異常
- 精胺丁二酸酵素缺乏症 疾病介紹及衛教
精胺丁二酸酵素缺乏症是一種尿素循環代謝異常疾病,致病原因為第7號染色體上的ASL基因變異所致,造成精胺琥珀水解酵素活性缺乏或功能降低,無法將精氨基琥珀酸轉化為精氨酸,導致尿素循環功能障礙。
- 異染性腦白質退化症 疾病介紹及衛教
異染性腦白質退化症是一種中樞神經系統疾病,致病原因為第22號染色體上的ARSA基因突變所致,使得分解硫酸鹽-醣脂的酵素arylsulfatase A缺乏或功能異常,導致硫酸鹽-醣脂逐漸累積在神經系統而損害神經。
- PRRT2相關陣發性運動障礙 疾病介紹及衛教
PRRT2相關陣發性運動障礙包括陣發性動作型運動不良症以及良性家族性嬰兒癲癇症,致病原因為第16號染色體上的PRRT2基因變異所致。PRRT2基因編碼富含脯胺酸的跨膜蛋白,存在於大腦的神經元中
- 先天性痛不敏感症合併無汗症 疾病介紹及衛教
先天性痛不敏感症合併無汗症,致病原因為第1號染色體上的NTRK1基因變異所致,轉錄出異常的NTRK1蛋白,造成無法與神經生長因子β(NGFβ)結合,影響細胞之間的信號傳遞功能,導致感覺神經元細胞逐漸凋亡
- 肌無力症候群第10型 疾病介紹及衛教
肌無力症候群第10型是一種神經肌肉傳導異常的疾病,致病原因為位在第4號染色體上的DOK7基因變異所致。此基因製造的蛋白質與神經肌肉突觸的形成及維持有
- 原發性先天性青光眼 疾病介紹及衛教
原發性先天性青光眼,或是原發性嬰幼兒型青光眼是一種罕見的遺傳性眼部疾病,常見的致病原因為第2號染色體上CYP1B1基因變異所致,造成細胞色素P450蛋白發生變異,影響眼睛內部睫狀體等結構的發
- 家族腎視網膜營養不良第6型 疾病介紹及衛教
家族腎視網膜營養不良症是一種罕見的影響身體多個器官的遺傳疾病,其臨床特徵是腎臟及眼睛疾病所組合而成。其中家族腎視網膜營養不良症第6型,致病原因為第12號染色體上的CEP290基因突變所致,基因變異導致合成初級纖毛細胞結構的蛋白質異常
- 法布瑞氏症 疾病介紹及衛教
法布瑞氏症是一種罕見的遺傳性溶小體儲積症,致病原因為X染色體上的GLA基因變異所致,此基因主要負責製造α-galactosidase(簡稱α-GAL)酵素。當體內缺乏此酵素時,會使得醣脂質,尤其是globotriaosylceramide(簡稱GL-3或Gb-3)無法被代謝
- 視網膜色素病變第59/第76型 疾病介紹及衛教
視網膜色素病變是一種遺傳性眼科疾病,依照變異基因不同區分型別,其中第 59型致病原因為第1號染色體DHDDS基因變異所致,導致感光受體醣蛋白(包括視紫紅質)醣基化作用異常,造成視網膜功能異常
- 先天性甲狀腺機能低下症第1型 疾病介紹及衛教
先天性甲狀腺機能低下症是一種常見的先天性代謝異常疾病,大致可歸類為甲狀腺發育不全或是甲狀腺激素生成異常而造成,其中,先天性甲狀腺機能低下症第1型致病原因為第14號染色體上的TSHR基因發生變異所致
- 腎病症候群第1型 疾病介紹及衛教
腎病症候群第1型是一種罕見的遺傳性腎臟疾病,致病原因為第19號染色體上的NPHS1基因變異所致。NPHS1基因會製造出nephrin蛋白質,此蛋白質具有細胞傳遞的功能,位於腎小球中兩個足細胞(podocyte)之間的區域,稱為狹縫隔膜
- 高鳥胺酸血症-高氨血症-高瓜胺酸血症症候群 疾病介紹及衛教
高鳥胺酸血症-高氨血症-高瓜胺酸血症症候群是一種很少見的遺傳性代謝異常疾病,致病原因為位於第13號染色體上的SLC25A15基因變異所致,使得尿素循環代謝過程中的運輸酶功能缺損,導致鳥胺酸經由運輸酶進入粒線體代謝途徑異常
- 阿爾斯特倫症候群 疾病介紹及衛教
阿爾斯特倫症候群是一種多症狀疾病,致病原因為位於第2號染色體上的ALMS1基因變異所致,導致調控纖毛形成和維持的蛋白受到影響,由於此蛋白存在於多數組織中,一旦基因發生突變將影響全身各部位的功能,包括視力、聽覺、心臟、腎臟、肺和肝臟等
- 視網膜色素病變第37型/增強型S圓錐症候群 疾病介紹及衛教
視網膜色素病變第37型/增強型S-圓錐症候群是一種遺傳性眼科疾病,致病原因為位於第15號染色體上的NR2E3基因變異所致,使得眼球內的感光受體異常,或是導致長波長的L/M(紅色/綠色)錐體功能降低,以及短波長S(藍色)錐體功能增強
- 尼曼匹克症 疾病介紹及衛教
尼曼匹克症是一種脂質代謝異常疾病,依照臨床症狀與基因分成A型、B型、C1型和C2型。其中A型與B型的致病原因為第11號染色體上的SMPD1基因變異所致,此基因變異造成神經鞘磷脂酶(sphingomyelinase)活性不足或缺乏
- 黏多醣症 疾病介紹及衛教
黏多醣症是一種遺傳性代謝疾病,依照臨床症狀和致病基因不同區分為7型,由IDUA、IDS、HGSNAT、GLB1、ARSB等基因調控,當基因發生變異時,會造成黏多醣水解酶缺乏或功能缺失。黏多醣又稱醣胺聚醣存在於身體內各個細胞中
- 體染色體隱性遺傳魚鱗癬第1型 疾病介紹及衛教
魚鱗癬(Ichthyosis)是一種表皮角質化異常疾病,依照致病比例及基因不同而分成多種型別,目前已知至少有20種型別,其中,遺傳性魚鱗癬第1型是一種層狀魚鱗癬(Lamellar ichthyosis, LI),致病原因為第14號染色體上的TGM1基因變異所致
- Joubert氏症候群(家族性小腦蚓部發育不全) 第2型/第5型 疾病介紹及衛教
Joubert氏症候群是一種多症狀疾病,依照致病比例及基因不同而分成多種型別,其中第2型的致病原因為第11號染色體上的TMEM216基因變異所致,約佔Joubert氏症候群患者中的2~3%;第5型的致病原因為第12號染色體上的CEP290基因變異所致,約佔Joubert氏症候群患者中的7~10%
- 肉鹼棕櫚醯基轉移酶缺乏症第2型 疾病介紹及衛教
肉鹼棕櫚醯基轉移酶缺乏症第2型是一種遺傳性代謝異常疾病。致病原因為位於第1號染色體上的CPT2基因變異所致,造成肉鹼棕櫚醯基轉移酶酵素活性降低。當酵素活性不足,肉鹼無法和長鏈脂肪酸分離,脂肪酸無法進一步被代謝為能量,而能量不足將導致低酮酸性低血糖、肌肉痛、肌無力等症狀
- Branchiootorenal症候群 疾病介紹及衛教
Branchiootorenal症候群是一種頸部、耳部以及腎臟發育異常的疾病。主要由3個基因變異所致,分別是EYA1, SIX5及SIX1基因;EYA1基因變異會造成Branchiootorenal症候群第1型,SIX5基因變異會造成Branchiootorenal症候群第2型,SIX1基因變異會造成Branchiootorenal症候群第3型。
- 高膽紅素血症 疾病介紹及衛教
高膽紅素血症,致病原因為第2號染色體上UGT1A1基因變異所致,使得肝臟細胞中膽紅素葡萄糖醛酸基轉移酵素含量降低或活性不足,造成未結合型膽紅素清除速率較差,導致膽紅素較高,患者的皮膚和眼白會出現黃疸症狀
- 3-β-羥基類固醇脫氫酶缺乏引起之先天性腎上腺增生症第2型 疾病介紹及衛教
3-β-羥基類固醇脫氫酶缺乏引起之先天性腎上腺增生症第2型,致病原因為第1號染色體上的HSD3B2基因變異所致,使得參與合成腎上腺皮質激素的酵素(3β-羥基類固醇脫氫酶)缺少
- 巴德-畢德氏症候群 疾病介紹及衛教
巴德-畢德氏症候群為一種體染色體隱性遺傳疾病,大多由BBS相關基因變異所致,依照致病比例及遺傳基因不同而分多種型別,目前已知有26種型別,較常見為以下4種型別
- 梅克爾症候群第4型 疾病介紹及衛教
梅克爾症候群是一種罕見的影響身體多個器官的遺傳疾病,依照致病比例及基因不同而分成多種型別,第4型的致病原因為第12號染色體上的CEP290基因變異所致
- 先天性醣基化障礙第Ia、Ib、Ic型 疾病介紹及衛教
先天性醣基化障礙是一種醣基化異常而造成的代謝疾病,依照突變基因不同區分型別,Ia型致病原因為第16號染色體上的PMM2基因突變;Ib型致病原因為第15號染色體上的MPI基因突變
- 原發性高草酸尿症 疾病介紹及衛教
原發性高草酸尿症為一種代謝異常的遺傳性疾病,根據不同基因及發病時間分為3種型別,第1型致病原因為第2號染色體上的AGXT基因變異所致,第2型致病原因為第9號染色體上的GRHPR基因變異所致
- 遺傳性果糖不耐症 疾病介紹及衛教
遺傳性果糖不耐症是一種代謝異常的遺傳性疾病,致病原因為第9號染色體上的ALDOB基因變異所致,使得肝臟中的醛縮酶B(aldolase B)缺乏,無法分解轉化細胞中的果糖-1-磷酸(fructose-1-phosphate)為甘油醛(glyceraldehyde),導致無法產生葡萄糖和乳酸
- 體染色體隱性遺傳羊毛狀頭髮/毛髮稀疏症 疾病介紹及衛教
體染色體隱性遺傳羊毛狀頭髮/毛髮稀疏症是一種罕見的影響毛髮生長的遺傳性疾病,致病原因為位於第3號染色體上的LIPH基因變異所致,造成參與毛囊增生及分化細胞之蛋白製造異常
- 二氫嘧啶脫氫酶缺乏症 疾病介紹及衛教
二氫嘧啶脫氫酶缺乏症為一種遺傳性代謝疾病,致病原因為第1號染色體上DPYD基因發生變異所致,使得製造二氫嘧啶脫氫酶效率降低,影響尿嘧啶(Uracil)及胸腺嘧啶(Thymine)等核苷酸的分解作用,導致體液中的尿嘧啶及胸腺嘧啶過量
- 彈性纖維假黃瘤 疾病介紹及衛教
彈性纖維假黃瘤,致病原因為位於第16號染色體上的ABCC6基因變異所致,造成體內鈣和其他礦物質堆積於彈性組織中,產生漸進式的鈣化或礦化,使彈性纖維變性
- 腎上腺腦白質失養症 疾病介紹及衛教
腎上腺腦白質失養症是一種遺傳性代謝疾病,致病原因為X染色體上的ABCD1基因變異所致,導致製造腎上腺腦白質失養蛋白功能異常,造成無法代謝極長鏈脂肪酸
- 吉特曼症候群 疾病介紹及衛教
吉特曼症候群是一種與腎臟有關的代謝疾病,致病原因為第16號染色體上的SLC12A3基因突變,導致無法製造鈉離子(Na+)與氯離子(Cl-)的共同運輸膜蛋白質,造成腎臟遠曲小管中鈉和氯離子的再吸收能力下降
- 腦腱性黃瘤症 疾病介紹及衛教
腦腱性黃瘤症是一種罕見的脂質代謝異常疾病,致病原因為第2對染色體上CYP27A1基因突變,導致固醇27-羥化酶功能異常,無法提供鵝去氧膽酸來有效分解膽固醇,造成脂質中的膽甾烷醇和膽固醇以黃色瘤的形式堆積在體內多個器官
- 17-α-羥化酶缺乏引起之先天性腎上腺增生症 疾病介紹及衛教
17-α-羥化酶缺乏引起之先天性腎上腺增生症,致病原因為位於第10號染色體上的CYP17A1基因突變所致,使得參與合成腎上腺皮質激素的酵素(17-α-羥化酶)缺乏,該酵素缺乏將導致皮質醇(cortisol)、醛固酮(aldosterone)缺少及雄激素(androgen)生成過量。
- 極長鏈醯輔酶Α去氫酶缺乏症 疾病介紹及衛教
極長鏈醯輔酶Α去氫酶缺乏症為一種代謝異常疾病,致病原因為第17對染色體上的ACADVL基因突變所致,使得體內缺乏極長鏈醯輔酶Α去氫酶(VLCAD),無法分解極長鏈脂肪酸轉換成能量,因而阻斷酮體形成與抑制醣質新生,引起嗜睡和低血糖等症狀。
- Aicardi-Goutieres症候群 疾病介紹及衛教
Aicardi-Goutieres症候群是一種影響大腦、免疫系統和皮膚的疾病,依照致病比例及遺傳基因不同而分多種型別。第1型致病原因為第3號染色體上TREX1基因突變,約占23%;第3型致病原因為第11號染色體上RNASEH2C基因突變,約占12%;第5型致病原因為第20號染色體上SAMHD1基因突變,約占13%。
- 肢帶型肌肉失養症第2A型 疾病介紹及衛教
肢帶型肌肉失養症第2A型,致病原因為位於第15號染色體上的CAPN3基因發生突變所致,導致肌肉細胞內鈣離子活化的蛋白分解酶(Calpain-3)發生異常,進而影響正常骨骼肌功能。CAPN3基因帶因率約為0.3~0.9%。
- 3-羥基-3-甲基戊二酸尿症 疾病介紹及衛教
3-羥基-3-甲基戊二酸尿症是一種代謝異常疾病,致病原因為第1號染色體上的HMGCL基因突變,導致3-羥基-3-甲基戊二醯輔酶A裂解酶(HMG-CoA)缺乏,此裂解酶主要功能是將蛋白質的白胺酸(Leucine)分解,並產生可生成能量的酮體(Ketone body)。
- 中鏈醯輔酶Α去氫酶缺乏症 疾病介紹及衛教
中鏈醯輔酶Α去氫酶缺乏症,致病原因為第1號染色體上的ACADM基因變異,造成中鏈醯輔酶Α去氫酶生成異常,使脂肪酸無法順利代謝成能量,過多的脂肪酸堆積於體內會產生毒性,對大腦及神經系統造成傷害。ACADM基因帶因率約為1.5%。
- 3-甲基巴豆醯輔酵素羧化酵素缺乏症 疾病介紹及衛教
3-甲基巴豆醯輔酵素羧化酵素缺乏症是一種代謝異常疾病。因致病基因不同分為2種型別,第1型致病原因為第3對染色體上的MCCC1或稱MCCA基因突變,第2型致病原因為第5對染色體上的MCCC2或稱MCCB 基因突變。
- 亨丁頓氏舞蹈症 疾病介紹及衛教
亨丁頓氏舞蹈症是一種神經退化性疾病,致病原因為第4號染色體上HTT基因CAG三核苷酸序列異常擴增,造成腦部基底核的GABA神經傳導素缺乏,導致漸進式的神經退化,疾病特徵為肢體肌肉產生不自主的運動、智能退化以及精神方面的問題。
- 尤塞氏症候群第1型 疾病介紹及衛教
尤塞氏症候群第1型為隱性遺傳疾病,主要的臨床特徵為先天性雙側重度至極重度聽損,以及漸進性視力喪失問題。尤塞氏症候群第1型的相關基因至少有6個,其中最常見的是第11號染色體上的MYO7A基因,約佔53~70%
- 異戊酸血症 疾病介紹及衛教
異戊酸血症是一種胺基酸代謝異常疾病,致病原因為第15對染色體上的IVD基因突變,造成異戊醯輔酶A去氫酶發生缺陷,無法分解異戊酸,導致大量異戊酸堆積於體內,侵害人體的神經與造血系統。
- 瓜胺酸血症 疾病介紹及衛教
瓜胺酸血症是一種尿素循環代謝異常疾病,人體在代謝蛋白質和氨基酸後會產生有毒的血氨,需要經由肝臟進行尿素循環反應,將有毒性的血氨轉換成較無毒性的尿素,再經由尿液排除至體外,疾病依致病機轉分為兩大類。
- 血友病 疾病介紹及衛教
血友病為一種先天性血液凝固異常的遺傳疾病,因為基因缺陷而造成血液中凝血因子的缺乏,導致身體的凝血機轉無法正常運作。
- 天冬氨酰葡萄糖胺尿症 疾病介紹及衛教
天冬氨酰葡萄糖胺尿症致病原因為位於第4號染色體上的AGA基因突變,導致溶小體中缺乏天冬氨酰葡萄糖苷酶,進而阻斷醣蛋白的正常分解而大量堆積在溶小體內。
- 低磷酸酯酶症 疾病介紹及衛教
低磷酸酯酶症是一種影響骨骼及牙齒發育的疾病,其致病原因為第1號染色體的ALPL基因發生突變,導致非特異性組織鹼性磷酸酶功能異常,無法參與骨骼組織礦化的過程,故無法形成堅硬的骨骼及牙齒。
- GM1神經節苷脂儲積症 疾病介紹及衛教
神經節苷脂儲積症為一代謝性遺傳疾病,致病原因為第3號染色體上的GLB1基因突變,使得溶小體中β-galactosidase酵素功能不良,造成無法分解GM1神經節苷脂,進而堆積於組織及器官中,影響大腦和脊髓神經細胞。
- 戊二酸血症第2型 疾病介紹及衛教
戊二酸血症第2型是一種脂肪酸氧化過程異常疾病,其致病原因為ETFA、ETFB或ETFDH基因變異,造成體內無法分解脂肪、蛋白質以及膽鹼,使得血液和組織變得過於酸性,形成代謝性酸中毒。
- 戊二酸血症第1型 疾病介紹及衛教
戊二酸血症第1型是一種胺基酸代謝異常疾病,其致病原因為GCDH基因變異,使戊二基輔酶A去氫酵素異常,使得有毒的代謝中間產物戊二酸過量堆積,導致日後生理發育障礙與智能遲緩。
- 甲狀腺激素生成障礙 疾病介紹及衛教
甲狀腺是人體中重要的內分泌器官之一,主要維持體內新陳代謝功能,若發育中的幼童缺少甲狀腺素,會造成生長遲緩及智力不足。若當甲狀腺激素生成之相關基因發生變異時,會造成甲狀腺合成途徑出現問題,導致甲狀腺激素缺少而致病。
- 多發性羧化酶缺乏症 疾病介紹及衛教
多發性羧化酶缺乏症,其致病原因為HLCS基因突變,使全羧化酶合成酶缺乏,全羧化酶合成酶可以和生物素結合去幫助分解蛋白質、脂質及碳水化合物,一旦HLCS基因產生致病突變,就會降低此酵素結合生物素的能力,造成多種羧化酶功能異常,阻止營養物質的分解,破壞細胞功能,而造成症狀表現。
- 丙酸血症 疾病介紹及衛教
丙酸血症為先天性代謝異常疾病,致病原因為PCCA或PCCB基因發生變異所致,使得丙醯基輔酵素A羧化酵素功能異常,導致大量堆積丙酸於體內發生代謝性酮酸中毒。
- 體染色體隱性遺傳多囊性腎臟病 疾病介紹及衛教
體染色體隱性遺傳多囊性腎臟病,其致病原因為PKHD1基因突變,造成嬰兒或幼童的肝臟及腎臟出現囊泡與纖維化特徵,嚴重情況下需做血液透析或進行腎臟移植。
- 楓糖尿症 疾病介紹與衛教
楓糖尿症是特殊支鏈胺基酸代謝異常疾病,致病原因為BCKDHA、BCKDHB、DBT基因發生變異所致,這三個基因中任一個基因變異會降低支鏈甲型酮酸脫氫酵素,造成支鏈胺基酸大量堆積及產生毒性,不僅對腦細胞造成傷害,也會產生如楓樹糖漿的特殊體味。
- 范可尼氏貧血症 疾病介紹及衛教
范可尼氏貧血症為一種遺傳性骨髓造血功能異常的疾病,其相關基因與DNA修復有關,當基因發生缺陷時會導致造血幹細胞異常凋亡,無法分化為成熟的血球細胞,造成骨髓造血功能失常,另外患者可能會有急性白血病或其他癌症等症狀。
- 高雪氏症 疾病介紹及衛教
高雪氏症是一種溶小體神經脂質儲積症,致病原因為第1號染色體上的GBA基因突變,使得葡萄糖腦甘脂酵素活性降低,造成代謝物異常堆積在肝、骨髓、脾臟及腦部等處,進而導致器官損傷或智力減退等症狀。
- 裘馨氏/貝克氏肌肉萎縮症 疾病介紹及衛教
裘馨氏/貝克氏肌肉萎縮症是一種進行性肌肉無力及萎縮之遺傳性疾病,其致病原因為X染色體上DMD基因變異,造成肌縮蛋白生成異常,影響肌肉細胞的完整性,導致肌肉細胞逐漸萎縮及壞死。
- 苯酮尿症 疾病介紹及衛教
苯酮尿症是一種代謝異常的遺傳性疾病,因致病機轉不同,可分為兩大型別,PAH和BH4缺乏型,因苯丙胺酸堆積無法正常代謝,會造成神經系統傷害,若無早期發現及治療,患者會有發育不良及智能障礙等症狀。
- 瓦登伯革氏症候群第4A型疾病介紹及衛教
瓦登伯革氏症候群又被稱為藍眼珠症候群,患者的眼珠虹膜異色,但視力並未受到影響,易合併聽障及長期便秘等問題。
- 遺傳性球形紅血球增多症第4型疾病介紹及衛教
遺傳性球形紅血球增多症是由調控紅血球細胞膜蛋白基因異常所引起的遺傳性疾病,此疾病兩大特徵為紅血球不足及脾臟中過多的紅血球。
- COL2A1 (Collagen Type II Alpha 1 Chain) 基因介紹
第2型膠原蛋白又稱軟骨膠原蛋白,主要參與軟骨細胞合成,主要存在的區域為軟骨組織、眼球玻璃體內、內耳及脊椎骨之間的椎間盤中心(髓核)等部位。當COL2A1基因發生變異時,就會造成軟骨發育不良,而導致臨床症狀出現。
- Nonaka肌病變 疾病介紹與衛教
Nonaka肌病變致病原因為GNE基因發生變異所致,使得細胞表面的唾液酸降低,造成細胞移動、附著及訊號傳遞等受到影響,導致漸進性的遠端肌無力。
- 球細胞腦白質失養症 疾病介紹與衛教
球細胞腦白質失養症其致病原因為GALC基因變異所致,使得半乳糖腦苷酯脢酵素活性缺乏無法正常代謝,進而大量堆積在神經細胞內,引發神經學特徵與症狀。
- 史黛氏症 疾病介紹及衛教
史黛氏症是一種罕見的遺傳性疾病,其致病原因是SBDS基因發生變異,使得參與製造蛋白質的核醣體異常,會影響骨髓、胰臟及骨骼等器官發育。
- 原發性肉鹼缺乏症疾病介紹與衛教
原發性肉鹼缺乏症為一種脂肪酸氧化異常疾病,患者因體內肉鹼缺乏,使得無法有效利用脂肪酸來產生能量,也無法產生足夠的酮體為大腦使用,導致可能出現心肌、腦神經、肌肉骨骼等方面出現病變。
- 肝醣儲積症第1型疾病介紹及衛教
肝醣儲積症第1型是一種遺傳代謝疾病,使患者缺乏葡萄糖-6-磷酸酵素功能,造成肝醣無法順利轉化成葡萄糖,除了發生低血糖外,也會導致體內累積過多肝醣,長期下來會損害多重臟器功能。
- 半乳糖血症疾病介紹與衛教
半乳糖血症是一種醣類代謝異常的疾病,使得半乳糖轉變成葡萄糖的代謝途徑發生障礙,導致體內半乳糖的堆積,造成生長及發展遲緩、智力障礙、肝臟疾病及腎臟問題等。
- 囊腫纖維症(Cystic fibrosis, CF)疾病介紹及衛教
囊腫纖維症其致病原因為CFTR 基因突變所致,導致患者的呼吸道、消化道、胰臟及汗腺等外分泌腺體器官功能異常,造成分泌物無法順利排出體外,增加感染風險。
- 蠶豆症(G6PD缺乏症)疾病介紹及衛教
蠶豆症是一種很常見的先天性代謝異常疾病,因G6PD基因變異會導致紅血球內G6PD酵素活性減少,當碰到特定氧化物質時,易產生溶血、貧血等症狀。
- 家族性地中海熱(Familial Mediterranean fever, FMF)疾病介紹及衛教
家族性地中海熱其致病原因為MEFV基因突變所致,造成患者白血球中的比林蛋白活性降低,使得體內發炎反應無法正常調節,患者會反覆發燒、胸腹部和關節處疼痛等臨床症狀。
- 染色體隱性遺傳疾病-血鐵沉積症(血色素沉著病)
血鐵沉積症,又稱「血色素沉著病」是一種小腸黏膜對鐵吸收過多鐵的疾病。臨床特徵是肝臟、皮膚、胰腺、心臟、關節和垂體前葉過度儲存鐵,在未經治療的患者中,早期症狀包括:腹痛、虛弱、嗜睡、體重減輕、關節痛等。長期鐵的累積會造成身體臟器受損。
- 容易忽略的罕見疾病「威爾森氏症」
威爾森氏症是一種罕見的遺傳疾病,患者時常因為被忽略病因而延誤了診斷與治療,發病年齡多為六至四十歲;其臨床症狀和發病年齡有個體差異性,從孩童時期到青壯年都有可能發病,疾病嚴重度也有很大的差異。
- 不正常的抽搐行為?原來是罕見疾病 結節性硬化症
5歲大的奇奇看起來活潑健康,但就在某個假日,奇奇的爺爺帶著到公園玩的時候,玩著玩著爺爺發現奇奇突然出現不正常的抽搐行為…
- 馬凡氏症候群|及早診斷與治療 降低併發症的風險
患者常常給人的第一印象就是高高瘦瘦的且手腳也長,因為有這樣的特徵,常會提拔為運動校隊或選手,若在不知情的狀態下進行激烈運動或訓練時,往往會造成心臟負擔,嚴重時會猝死危及生命安全
- 肝醣儲積症第二型 龐貝氏症|父母健康 卻生下遺傳疾病寶寶?
肝醣儲積症第二型(Glycogen Storage Disease type II)又稱龐貝氏症(Pompe Disease);患者最主要臨床症狀為肌肉相關問題,以肌肉張力降低、無力為主。龐貝氏症依照發病時間點可分為嬰兒型和晚發型兩種。
- 眼睛也會長腫瘤? 當心兒童常見眼癌影響寶貝視力
依據中華民國兒童癌症基金會統計,「視網膜母細胞瘤」為台灣十大兒童癌症之一,亦是兒童最常見的眼科癌症。
- 視網膜母細胞瘤
為一種較為罕見的眼部腫瘤疾病,是在兒童眼睛腫瘤中最常見的原發性惡性腫瘤,大部份發病的年齡介於0~5歲的嬰幼兒中
- 眼睛皮膚白化症 Oculocutaneous Albinism
眼睛皮膚白化症是一種影響皮膚、頭髮和眼睛著色(色素沉著)的遺傳疾病,其致病原因為相關基因變異導致所製造的蛋白有缺陷,使得色素細胞製造出的黑色素減少
- 矮小基因缺失症(SHOX Deficiency)
造成身材矮小的原因,除了年齡、營養、壓力、生活型態之外,研究指出SHOX(矮小基因)基因也會導致嚴重影響
- 進行性神經性腓骨萎縮症/恰克-馬利-杜斯氏症/遺傳性運動感覺神經病變Charcot Marie Tooth Disease
發病時間從兒童時期至成人時期皆有可能,患者在臨床症狀上通常為對稱性的,從手部和腿部的遠端運動神經開始病變,而導致手掌與腳掌的肌肉逐漸無力與開始萎縮,讓患者在行動與生活造成困難與不便
- 認識先天性腎上腺增生症
是由於類固醇-21 羥化脢(Steroid 21-hydroxylase, CYP21A2)的基因變異所造成,因此基因變異導致先天缺少某些製造腎上腺皮質素的酵素,進而造成腎上腺疾病;依酵素缺乏程度可能有程度不一的男性化徵象,嚴重者會有鈉和水分不平衡等問題
- 寶寶患有先天性腎上腺增生症怎麼辦?
先天性腎上腺增生症的酵素數值異常,在臨床上嚴重者可能會有嗜睡嘔吐、或是性器官混淆的狀況,輕微者可能會有性早熟情況發生
- 裘馨氏肌肉萎縮症
是肌肉萎縮症中最常見的一種,根據歐美統計每十萬新生男嬰中有約20~30人患病,其致病原因是位於X染色體短臂Xp21的Dystrophin基因(DMD gene)有異常而造成的
- 人體發電廠失靈?罕病粒線體代謝異常是什麼?
粒線體基因突變大部份為遺傳性,若當粒線體缺陷時,就無法釋出足夠的能量,對於需要大量能量的器官就會產生病變
- 血液異常凝結?帶你認識蛋白質C缺乏症
蛋白質C缺乏症(Protein C deficiency)是一種會讓血液異常凝結風險增加的疾病,其致病原因為PROC基因變異使得製造蛋白質C的功能受影響而導致
- 蛋白質C缺乏症|新生兒PROC基因突變?恐致血管栓塞、猛爆性紫斑症
為什麼小華看起來好好的,卻有這個基因異常?在網路上查到關於蛋白質C缺乏症患者的臨床症狀,未來是不是都會出現在小華身上?
- 患者福音! 三種SMA脊髓性肌肉萎縮症用藥
目前美國FDA核准三種藥物可治療脊髓性肌肉萎縮症,可以改善患者的肌肉運動功能並且提升整體存活率
- 周歲寶寶想走路卻雙腳無力?原來是此遺傳疾病導致
李爸爸、李媽媽發現每當帶著倫倫站立的時候,倫倫的腳好像都沒什麼力氣。透過臨床診斷以及基因檢查,確定了倫倫罹患了脊髓性肌肉萎縮症
- 脊髓性肌肉萎縮症SMA知多少?帶因率與致病率皆高的遺傳性疾病
脊髓性肌肉萎縮症(Spinal Muscular Atrophy, SMA)是一種可能致命的遺傳性疾病。發病年齡從出生到成年皆有可能發生,每個人依照發病年齡以及疾病的嚴重程度,症狀會有很大的差異
- 健康超前部署 淺談基因檢測在現代醫療的應用
目前有越來越多的疾病被發現跟基因有關係。基因檢測在目前科學的應用有哪些呢?主要可以分為疾病診斷、預防醫學、精準醫療三個方面
- 家族性澱粉樣多發性神經病變之標靶治療:Onpattro、Vyndaqel
關於家族性澱粉樣多發性神經病變之標靶治療用藥分析
- 肢體無力、腕隧道症候群?認識家族性澱粉樣多發性神經病變
是一種罕見疾病,依不同的疾病類型與種族,其發生率與發病年齡也有所不同。臨床特徵可包括周圍感覺運動神經病變和自主神經病變
- 寶寶常跌倒、無法彎腰?小心是先天性肌失養症
肌肉細胞中某部分的蛋白質無法獲得適當營養,進而出現變性壞死或造成結構變化,因此出現肌肉力量降低的症狀
- 致命危險?認識遺傳性出血性血管擴張症
此疾病與血管生成及修補異常有關,非血液凝固問題或凝血因子缺乏所造成。患者通常有反覆性鼻出血的症狀,平均發病年齡為12歲
- 遺傳性出血性血管擴張症|流鼻血也會遺傳?透過寶寶基因檢測才發現誰是帶原者
芯芯媽媽想起自己舅舅經常性的流鼻血,但自己並沒有這樣的情況,有可能會是自己遺傳給女兒嗎?

 酪胺酸血症是一種遺傳性代謝疾病,依照突變基因不同區分型別;第1型的致病原因為第15號染色體上的FAH基因突變,造成蛋白質功能失常,無法將延胡索醯乙醯乙酸轉換成延胡索酸和乙醯乙酸
酪胺酸血症是一種遺傳性代謝疾病,依照突變基因不同區分型別;第1型的致病原因為第15號染色體上的FAH基因突變,造成蛋白質功能失常,無法將延胡索醯乙醯乙酸轉換成延胡索酸和乙醯乙酸 α1-抗胰蛋白酶缺乏症是一種遺傳性內分泌疾病,致病原因為第14號染色體上的SERPINA1基因突變,導致製造α1-抗胰蛋白酶(α-1 antitrypsin,AAT)功能異常或低
α1-抗胰蛋白酶缺乏症是一種遺傳性內分泌疾病,致病原因為第14號染色體上的SERPINA1基因突變,導致製造α1-抗胰蛋白酶(α-1 antitrypsin,AAT)功能異常或低 線狀體肌肉病變第2型是一種非進行性的先天性神經肌肉病變,致病原因為第2號染色體上的NEB基因變異所致,使得Nebulin蛋白製造不足或功能降低。
線狀體肌肉病變第2型是一種非進行性的先天性神經肌肉病變,致病原因為第2號染色體上的NEB基因變異所致,使得Nebulin蛋白製造不足或功能降低。 短鏈醯輔酶Α去氫酶缺乏症是一種脂肪酸代謝異常疾病,致病原因為第12號染色體上的ACADS基因變異所致,導致短鏈醯輔酶Α去氫酶(SCAD)缺乏或功能異常
短鏈醯輔酶Α去氫酶缺乏症是一種脂肪酸代謝異常疾病,致病原因為第12號染色體上的ACADS基因變異所致,導致短鏈醯輔酶Α去氫酶(SCAD)缺乏或功能異常 精胺丁二酸酵素缺乏症是一種尿素循環代謝異常疾病,致病原因為第7號染色體上的ASL基因變異所致,造成精胺琥珀水解酵素活性缺乏或功能降低,無法將精氨基琥珀酸轉化為精氨酸,導致尿素循環功能障礙。
精胺丁二酸酵素缺乏症是一種尿素循環代謝異常疾病,致病原因為第7號染色體上的ASL基因變異所致,造成精胺琥珀水解酵素活性缺乏或功能降低,無法將精氨基琥珀酸轉化為精氨酸,導致尿素循環功能障礙。 異染性腦白質退化症是一種中樞神經系統疾病,致病原因為第22號染色體上的ARSA基因突變所致,使得分解硫酸鹽-醣脂的酵素arylsulfatase A缺乏或功能異常,導致硫酸鹽-醣脂逐漸累積在神經系統而損害神經。
異染性腦白質退化症是一種中樞神經系統疾病,致病原因為第22號染色體上的ARSA基因突變所致,使得分解硫酸鹽-醣脂的酵素arylsulfatase A缺乏或功能異常,導致硫酸鹽-醣脂逐漸累積在神經系統而損害神經。 PRRT2相關陣發性運動障礙包括陣發性動作型運動不良症以及良性家族性嬰兒癲癇症,致病原因為第16號染色體上的PRRT2基因變異所致。PRRT2基因編碼富含脯胺酸的跨膜蛋白,存在於大腦的神經元中
PRRT2相關陣發性運動障礙包括陣發性動作型運動不良症以及良性家族性嬰兒癲癇症,致病原因為第16號染色體上的PRRT2基因變異所致。PRRT2基因編碼富含脯胺酸的跨膜蛋白,存在於大腦的神經元中 先天性痛不敏感症合併無汗症,致病原因為第1號染色體上的NTRK1基因變異所致,轉錄出異常的NTRK1蛋白,造成無法與神經生長因子β(NGFβ)結合,影響細胞之間的信號傳遞功能,導致感覺神經元細胞逐漸凋亡
先天性痛不敏感症合併無汗症,致病原因為第1號染色體上的NTRK1基因變異所致,轉錄出異常的NTRK1蛋白,造成無法與神經生長因子β(NGFβ)結合,影響細胞之間的信號傳遞功能,導致感覺神經元細胞逐漸凋亡 肌無力症候群第10型是一種神經肌肉傳導異常的疾病,致病原因為位在第4號染色體上的DOK7基因變異所致。此基因製造的蛋白質與神經肌肉突觸的形成及維持有
肌無力症候群第10型是一種神經肌肉傳導異常的疾病,致病原因為位在第4號染色體上的DOK7基因變異所致。此基因製造的蛋白質與神經肌肉突觸的形成及維持有 原發性先天性青光眼,或是原發性嬰幼兒型青光眼是一種罕見的遺傳性眼部疾病,常見的致病原因為第2號染色體上CYP1B1基因變異所致,造成細胞色素P450蛋白發生變異,影響眼睛內部睫狀體等結構的發
原發性先天性青光眼,或是原發性嬰幼兒型青光眼是一種罕見的遺傳性眼部疾病,常見的致病原因為第2號染色體上CYP1B1基因變異所致,造成細胞色素P450蛋白發生變異,影響眼睛內部睫狀體等結構的發 家族腎視網膜營養不良症是一種罕見的影響身體多個器官的遺傳疾病,其臨床特徵是腎臟及眼睛疾病所組合而成。其中家族腎視網膜營養不良症第6型,致病原因為第12號染色體上的CEP290基因突變所致,基因變異導致合成初級纖毛細胞結構的蛋白質異常
家族腎視網膜營養不良症是一種罕見的影響身體多個器官的遺傳疾病,其臨床特徵是腎臟及眼睛疾病所組合而成。其中家族腎視網膜營養不良症第6型,致病原因為第12號染色體上的CEP290基因突變所致,基因變異導致合成初級纖毛細胞結構的蛋白質異常 法布瑞氏症是一種罕見的遺傳性溶小體儲積症,致病原因為X染色體上的GLA基因變異所致,此基因主要負責製造α-galactosidase(簡稱α-GAL)酵素。當體內缺乏此酵素時,會使得醣脂質,尤其是globotriaosylceramide(簡稱GL-3或Gb-3)無法被代謝
法布瑞氏症是一種罕見的遺傳性溶小體儲積症,致病原因為X染色體上的GLA基因變異所致,此基因主要負責製造α-galactosidase(簡稱α-GAL)酵素。當體內缺乏此酵素時,會使得醣脂質,尤其是globotriaosylceramide(簡稱GL-3或Gb-3)無法被代謝 視網膜色素病變是一種遺傳性眼科疾病,依照變異基因不同區分型別,其中第 59型致病原因為第1號染色體DHDDS基因變異所致,導致感光受體醣蛋白(包括視紫紅質)醣基化作用異常,造成視網膜功能異常
視網膜色素病變是一種遺傳性眼科疾病,依照變異基因不同區分型別,其中第 59型致病原因為第1號染色體DHDDS基因變異所致,導致感光受體醣蛋白(包括視紫紅質)醣基化作用異常,造成視網膜功能異常 先天性甲狀腺機能低下症是一種常見的先天性代謝異常疾病,大致可歸類為甲狀腺發育不全或是甲狀腺激素生成異常而造成,其中,先天性甲狀腺機能低下症第1型致病原因為第14號染色體上的TSHR基因發生變異所致
先天性甲狀腺機能低下症是一種常見的先天性代謝異常疾病,大致可歸類為甲狀腺發育不全或是甲狀腺激素生成異常而造成,其中,先天性甲狀腺機能低下症第1型致病原因為第14號染色體上的TSHR基因發生變異所致 腎病症候群第1型是一種罕見的遺傳性腎臟疾病,致病原因為第19號染色體上的NPHS1基因變異所致。NPHS1基因會製造出nephrin蛋白質,此蛋白質具有細胞傳遞的功能,位於腎小球中兩個足細胞(podocyte)之間的區域,稱為狹縫隔膜
腎病症候群第1型是一種罕見的遺傳性腎臟疾病,致病原因為第19號染色體上的NPHS1基因變異所致。NPHS1基因會製造出nephrin蛋白質,此蛋白質具有細胞傳遞的功能,位於腎小球中兩個足細胞(podocyte)之間的區域,稱為狹縫隔膜 高鳥胺酸血症-高氨血症-高瓜胺酸血症症候群是一種很少見的遺傳性代謝異常疾病,致病原因為位於第13號染色體上的SLC25A15基因變異所致,使得尿素循環代謝過程中的運輸酶功能缺損,導致鳥胺酸經由運輸酶進入粒線體代謝途徑異常
高鳥胺酸血症-高氨血症-高瓜胺酸血症症候群是一種很少見的遺傳性代謝異常疾病,致病原因為位於第13號染色體上的SLC25A15基因變異所致,使得尿素循環代謝過程中的運輸酶功能缺損,導致鳥胺酸經由運輸酶進入粒線體代謝途徑異常 阿爾斯特倫症候群是一種多症狀疾病,致病原因為位於第2號染色體上的ALMS1基因變異所致,導致調控纖毛形成和維持的蛋白受到影響,由於此蛋白存在於多數組織中,一旦基因發生突變將影響全身各部位的功能,包括視力、聽覺、心臟、腎臟、肺和肝臟等
阿爾斯特倫症候群是一種多症狀疾病,致病原因為位於第2號染色體上的ALMS1基因變異所致,導致調控纖毛形成和維持的蛋白受到影響,由於此蛋白存在於多數組織中,一旦基因發生突變將影響全身各部位的功能,包括視力、聽覺、心臟、腎臟、肺和肝臟等 視網膜色素病變第37型/增強型S-圓錐症候群是一種遺傳性眼科疾病,致病原因為位於第15號染色體上的NR2E3基因變異所致,使得眼球內的感光受體異常,或是導致長波長的L/M(紅色/綠色)錐體功能降低,以及短波長S(藍色)錐體功能增強
視網膜色素病變第37型/增強型S-圓錐症候群是一種遺傳性眼科疾病,致病原因為位於第15號染色體上的NR2E3基因變異所致,使得眼球內的感光受體異常,或是導致長波長的L/M(紅色/綠色)錐體功能降低,以及短波長S(藍色)錐體功能增強 尼曼匹克症是一種脂質代謝異常疾病,依照臨床症狀與基因分成A型、B型、C1型和C2型。其中A型與B型的致病原因為第11號染色體上的SMPD1基因變異所致,此基因變異造成神經鞘磷脂酶(sphingomyelinase)活性不足或缺乏
尼曼匹克症是一種脂質代謝異常疾病,依照臨床症狀與基因分成A型、B型、C1型和C2型。其中A型與B型的致病原因為第11號染色體上的SMPD1基因變異所致,此基因變異造成神經鞘磷脂酶(sphingomyelinase)活性不足或缺乏 黏多醣症是一種遺傳性代謝疾病,依照臨床症狀和致病基因不同區分為7型,由IDUA、IDS、HGSNAT、GLB1、ARSB等基因調控,當基因發生變異時,會造成黏多醣水解酶缺乏或功能缺失。黏多醣又稱醣胺聚醣存在於身體內各個細胞中
黏多醣症是一種遺傳性代謝疾病,依照臨床症狀和致病基因不同區分為7型,由IDUA、IDS、HGSNAT、GLB1、ARSB等基因調控,當基因發生變異時,會造成黏多醣水解酶缺乏或功能缺失。黏多醣又稱醣胺聚醣存在於身體內各個細胞中 魚鱗癬(Ichthyosis)是一種表皮角質化異常疾病,依照致病比例及基因不同而分成多種型別,目前已知至少有20種型別,其中,遺傳性魚鱗癬第1型是一種層狀魚鱗癬(Lamellar ichthyosis, LI),致病原因為第14號染色體上的TGM1基因變異所致
魚鱗癬(Ichthyosis)是一種表皮角質化異常疾病,依照致病比例及基因不同而分成多種型別,目前已知至少有20種型別,其中,遺傳性魚鱗癬第1型是一種層狀魚鱗癬(Lamellar ichthyosis, LI),致病原因為第14號染色體上的TGM1基因變異所致 Joubert氏症候群是一種多症狀疾病,依照致病比例及基因不同而分成多種型別,其中第2型的致病原因為第11號染色體上的TMEM216基因變異所致,約佔Joubert氏症候群患者中的2~3%;第5型的致病原因為第12號染色體上的CEP290基因變異所致,約佔Joubert氏症候群患者中的7~10%
Joubert氏症候群是一種多症狀疾病,依照致病比例及基因不同而分成多種型別,其中第2型的致病原因為第11號染色體上的TMEM216基因變異所致,約佔Joubert氏症候群患者中的2~3%;第5型的致病原因為第12號染色體上的CEP290基因變異所致,約佔Joubert氏症候群患者中的7~10% 肉鹼棕櫚醯基轉移酶缺乏症第2型是一種遺傳性代謝異常疾病。致病原因為位於第1號染色體上的CPT2基因變異所致,造成肉鹼棕櫚醯基轉移酶酵素活性降低。當酵素活性不足,肉鹼無法和長鏈脂肪酸分離,脂肪酸無法進一步被代謝為能量,而能量不足將導致低酮酸性低血糖、肌肉痛、肌無力等症狀
肉鹼棕櫚醯基轉移酶缺乏症第2型是一種遺傳性代謝異常疾病。致病原因為位於第1號染色體上的CPT2基因變異所致,造成肉鹼棕櫚醯基轉移酶酵素活性降低。當酵素活性不足,肉鹼無法和長鏈脂肪酸分離,脂肪酸無法進一步被代謝為能量,而能量不足將導致低酮酸性低血糖、肌肉痛、肌無力等症狀 Branchiootorenal症候群是一種頸部、耳部以及腎臟發育異常的疾病。主要由3個基因變異所致,分別是EYA1, SIX5及SIX1基因;EYA1基因變異會造成Branchiootorenal症候群第1型,SIX5基因變異會造成Branchiootorenal症候群第2型,SIX1基因變異會造成Branchiootorenal症候群第3型。
Branchiootorenal症候群是一種頸部、耳部以及腎臟發育異常的疾病。主要由3個基因變異所致,分別是EYA1, SIX5及SIX1基因;EYA1基因變異會造成Branchiootorenal症候群第1型,SIX5基因變異會造成Branchiootorenal症候群第2型,SIX1基因變異會造成Branchiootorenal症候群第3型。 高膽紅素血症,致病原因為第2號染色體上UGT1A1基因變異所致,使得肝臟細胞中膽紅素葡萄糖醛酸基轉移酵素含量降低或活性不足,造成未結合型膽紅素清除速率較差,導致膽紅素較高,患者的皮膚和眼白會出現黃疸症狀
高膽紅素血症,致病原因為第2號染色體上UGT1A1基因變異所致,使得肝臟細胞中膽紅素葡萄糖醛酸基轉移酵素含量降低或活性不足,造成未結合型膽紅素清除速率較差,導致膽紅素較高,患者的皮膚和眼白會出現黃疸症狀 3-β-羥基類固醇脫氫酶缺乏引起之先天性腎上腺增生症第2型,致病原因為第1號染色體上的HSD3B2基因變異所致,使得參與合成腎上腺皮質激素的酵素(3β-羥基類固醇脫氫酶)缺少
3-β-羥基類固醇脫氫酶缺乏引起之先天性腎上腺增生症第2型,致病原因為第1號染色體上的HSD3B2基因變異所致,使得參與合成腎上腺皮質激素的酵素(3β-羥基類固醇脫氫酶)缺少 巴德-畢德氏症候群為一種體染色體隱性遺傳疾病,大多由BBS相關基因變異所致,依照致病比例及遺傳基因不同而分多種型別,目前已知有26種型別,較常見為以下4種型別
巴德-畢德氏症候群為一種體染色體隱性遺傳疾病,大多由BBS相關基因變異所致,依照致病比例及遺傳基因不同而分多種型別,目前已知有26種型別,較常見為以下4種型別 梅克爾症候群是一種罕見的影響身體多個器官的遺傳疾病,依照致病比例及基因不同而分成多種型別,第4型的致病原因為第12號染色體上的CEP290基因變異所致
梅克爾症候群是一種罕見的影響身體多個器官的遺傳疾病,依照致病比例及基因不同而分成多種型別,第4型的致病原因為第12號染色體上的CEP290基因變異所致 先天性醣基化障礙是一種醣基化異常而造成的代謝疾病,依照突變基因不同區分型別,Ia型致病原因為第16號染色體上的PMM2基因突變;Ib型致病原因為第15號染色體上的MPI基因突變
先天性醣基化障礙是一種醣基化異常而造成的代謝疾病,依照突變基因不同區分型別,Ia型致病原因為第16號染色體上的PMM2基因突變;Ib型致病原因為第15號染色體上的MPI基因突變 原發性高草酸尿症為一種代謝異常的遺傳性疾病,根據不同基因及發病時間分為3種型別,第1型致病原因為第2號染色體上的AGXT基因變異所致,第2型致病原因為第9號染色體上的GRHPR基因變異所致
原發性高草酸尿症為一種代謝異常的遺傳性疾病,根據不同基因及發病時間分為3種型別,第1型致病原因為第2號染色體上的AGXT基因變異所致,第2型致病原因為第9號染色體上的GRHPR基因變異所致 遺傳性果糖不耐症是一種代謝異常的遺傳性疾病,致病原因為第9號染色體上的ALDOB基因變異所致,使得肝臟中的醛縮酶B(aldolase B)缺乏,無法分解轉化細胞中的果糖-1-磷酸(fructose-1-phosphate)為甘油醛(glyceraldehyde),導致無法產生葡萄糖和乳酸
遺傳性果糖不耐症是一種代謝異常的遺傳性疾病,致病原因為第9號染色體上的ALDOB基因變異所致,使得肝臟中的醛縮酶B(aldolase B)缺乏,無法分解轉化細胞中的果糖-1-磷酸(fructose-1-phosphate)為甘油醛(glyceraldehyde),導致無法產生葡萄糖和乳酸 體染色體隱性遺傳羊毛狀頭髮/毛髮稀疏症是一種罕見的影響毛髮生長的遺傳性疾病,致病原因為位於第3號染色體上的LIPH基因變異所致,造成參與毛囊增生及分化細胞之蛋白製造異常
體染色體隱性遺傳羊毛狀頭髮/毛髮稀疏症是一種罕見的影響毛髮生長的遺傳性疾病,致病原因為位於第3號染色體上的LIPH基因變異所致,造成參與毛囊增生及分化細胞之蛋白製造異常 二氫嘧啶脫氫酶缺乏症為一種遺傳性代謝疾病,致病原因為第1號染色體上DPYD基因發生變異所致,使得製造二氫嘧啶脫氫酶效率降低,影響尿嘧啶(Uracil)及胸腺嘧啶(Thymine)等核苷酸的分解作用,導致體液中的尿嘧啶及胸腺嘧啶過量
二氫嘧啶脫氫酶缺乏症為一種遺傳性代謝疾病,致病原因為第1號染色體上DPYD基因發生變異所致,使得製造二氫嘧啶脫氫酶效率降低,影響尿嘧啶(Uracil)及胸腺嘧啶(Thymine)等核苷酸的分解作用,導致體液中的尿嘧啶及胸腺嘧啶過量 彈性纖維假黃瘤,致病原因為位於第16號染色體上的ABCC6基因變異所致,造成體內鈣和其他礦物質堆積於彈性組織中,產生漸進式的鈣化或礦化,使彈性纖維變性
彈性纖維假黃瘤,致病原因為位於第16號染色體上的ABCC6基因變異所致,造成體內鈣和其他礦物質堆積於彈性組織中,產生漸進式的鈣化或礦化,使彈性纖維變性 腎上腺腦白質失養症是一種遺傳性代謝疾病,致病原因為X染色體上的ABCD1基因變異所致,導致製造腎上腺腦白質失養蛋白功能異常,造成無法代謝極長鏈脂肪酸
腎上腺腦白質失養症是一種遺傳性代謝疾病,致病原因為X染色體上的ABCD1基因變異所致,導致製造腎上腺腦白質失養蛋白功能異常,造成無法代謝極長鏈脂肪酸 吉特曼症候群是一種與腎臟有關的代謝疾病,致病原因為第16號染色體上的SLC12A3基因突變,導致無法製造鈉離子(Na+)與氯離子(Cl-)的共同運輸膜蛋白質,造成腎臟遠曲小管中鈉和氯離子的再吸收能力下降
吉特曼症候群是一種與腎臟有關的代謝疾病,致病原因為第16號染色體上的SLC12A3基因突變,導致無法製造鈉離子(Na+)與氯離子(Cl-)的共同運輸膜蛋白質,造成腎臟遠曲小管中鈉和氯離子的再吸收能力下降 腦腱性黃瘤症是一種罕見的脂質代謝異常疾病,致病原因為第2對染色體上CYP27A1基因突變,導致固醇27-羥化酶功能異常,無法提供鵝去氧膽酸來有效分解膽固醇,造成脂質中的膽甾烷醇和膽固醇以黃色瘤的形式堆積在體內多個器官
腦腱性黃瘤症是一種罕見的脂質代謝異常疾病,致病原因為第2對染色體上CYP27A1基因突變,導致固醇27-羥化酶功能異常,無法提供鵝去氧膽酸來有效分解膽固醇,造成脂質中的膽甾烷醇和膽固醇以黃色瘤的形式堆積在體內多個器官 17-α-羥化酶缺乏引起之先天性腎上腺增生症,致病原因為位於第10號染色體上的CYP17A1基因突變所致,使得參與合成腎上腺皮質激素的酵素(17-α-羥化酶)缺乏,該酵素缺乏將導致皮質醇(cortisol)、醛固酮(aldosterone)缺少及雄激素(androgen)生成過量。
17-α-羥化酶缺乏引起之先天性腎上腺增生症,致病原因為位於第10號染色體上的CYP17A1基因突變所致,使得參與合成腎上腺皮質激素的酵素(17-α-羥化酶)缺乏,該酵素缺乏將導致皮質醇(cortisol)、醛固酮(aldosterone)缺少及雄激素(androgen)生成過量。 極長鏈醯輔酶Α去氫酶缺乏症為一種代謝異常疾病,致病原因為第17對染色體上的ACADVL基因突變所致,使得體內缺乏極長鏈醯輔酶Α去氫酶(VLCAD),無法分解極長鏈脂肪酸轉換成能量,因而阻斷酮體形成與抑制醣質新生,引起嗜睡和低血糖等症狀。
極長鏈醯輔酶Α去氫酶缺乏症為一種代謝異常疾病,致病原因為第17對染色體上的ACADVL基因突變所致,使得體內缺乏極長鏈醯輔酶Α去氫酶(VLCAD),無法分解極長鏈脂肪酸轉換成能量,因而阻斷酮體形成與抑制醣質新生,引起嗜睡和低血糖等症狀。 Aicardi-Goutieres症候群是一種影響大腦、免疫系統和皮膚的疾病,依照致病比例及遺傳基因不同而分多種型別。第1型致病原因為第3號染色體上TREX1基因突變,約占23%;第3型致病原因為第11號染色體上RNASEH2C基因突變,約占12%;第5型致病原因為第20號染色體上SAMHD1基因突變,約占13%。
Aicardi-Goutieres症候群是一種影響大腦、免疫系統和皮膚的疾病,依照致病比例及遺傳基因不同而分多種型別。第1型致病原因為第3號染色體上TREX1基因突變,約占23%;第3型致病原因為第11號染色體上RNASEH2C基因突變,約占12%;第5型致病原因為第20號染色體上SAMHD1基因突變,約占13%。 肢帶型肌肉失養症第2A型,致病原因為位於第15號染色體上的CAPN3基因發生突變所致,導致肌肉細胞內鈣離子活化的蛋白分解酶(Calpain-3)發生異常,進而影響正常骨骼肌功能。CAPN3基因帶因率約為0.3~0.9%。
肢帶型肌肉失養症第2A型,致病原因為位於第15號染色體上的CAPN3基因發生突變所致,導致肌肉細胞內鈣離子活化的蛋白分解酶(Calpain-3)發生異常,進而影響正常骨骼肌功能。CAPN3基因帶因率約為0.3~0.9%。 3-羥基-3-甲基戊二酸尿症是一種代謝異常疾病,致病原因為第1號染色體上的HMGCL基因突變,導致3-羥基-3-甲基戊二醯輔酶A裂解酶(HMG-CoA)缺乏,此裂解酶主要功能是將蛋白質的白胺酸(Leucine)分解,並產生可生成能量的酮體(Ketone body)。
3-羥基-3-甲基戊二酸尿症是一種代謝異常疾病,致病原因為第1號染色體上的HMGCL基因突變,導致3-羥基-3-甲基戊二醯輔酶A裂解酶(HMG-CoA)缺乏,此裂解酶主要功能是將蛋白質的白胺酸(Leucine)分解,並產生可生成能量的酮體(Ketone body)。 中鏈醯輔酶Α去氫酶缺乏症,致病原因為第1號染色體上的ACADM基因變異,造成中鏈醯輔酶Α去氫酶生成異常,使脂肪酸無法順利代謝成能量,過多的脂肪酸堆積於體內會產生毒性,對大腦及神經系統造成傷害。ACADM基因帶因率約為1.5%。
中鏈醯輔酶Α去氫酶缺乏症,致病原因為第1號染色體上的ACADM基因變異,造成中鏈醯輔酶Α去氫酶生成異常,使脂肪酸無法順利代謝成能量,過多的脂肪酸堆積於體內會產生毒性,對大腦及神經系統造成傷害。ACADM基因帶因率約為1.5%。 3-甲基巴豆醯輔酵素羧化酵素缺乏症是一種代謝異常疾病。因致病基因不同分為2種型別,第1型致病原因為第3對染色體上的MCCC1或稱MCCA基因突變,第2型致病原因為第5對染色體上的MCCC2或稱MCCB 基因突變。
3-甲基巴豆醯輔酵素羧化酵素缺乏症是一種代謝異常疾病。因致病基因不同分為2種型別,第1型致病原因為第3對染色體上的MCCC1或稱MCCA基因突變,第2型致病原因為第5對染色體上的MCCC2或稱MCCB 基因突變。 亨丁頓氏舞蹈症是一種神經退化性疾病,致病原因為第4號染色體上HTT基因CAG三核苷酸序列異常擴增,造成腦部基底核的GABA神經傳導素缺乏,導致漸進式的神經退化,疾病特徵為肢體肌肉產生不自主的運動、智能退化以及精神方面的問題。
亨丁頓氏舞蹈症是一種神經退化性疾病,致病原因為第4號染色體上HTT基因CAG三核苷酸序列異常擴增,造成腦部基底核的GABA神經傳導素缺乏,導致漸進式的神經退化,疾病特徵為肢體肌肉產生不自主的運動、智能退化以及精神方面的問題。 尤塞氏症候群第1型為隱性遺傳疾病,主要的臨床特徵為先天性雙側重度至極重度聽損,以及漸進性視力喪失問題。尤塞氏症候群第1型的相關基因至少有6個,其中最常見的是第11號染色體上的MYO7A基因,約佔53~70%
尤塞氏症候群第1型為隱性遺傳疾病,主要的臨床特徵為先天性雙側重度至極重度聽損,以及漸進性視力喪失問題。尤塞氏症候群第1型的相關基因至少有6個,其中最常見的是第11號染色體上的MYO7A基因,約佔53~70% 異戊酸血症是一種胺基酸代謝異常疾病,致病原因為第15對染色體上的IVD基因突變,造成異戊醯輔酶A去氫酶發生缺陷,無法分解異戊酸,導致大量異戊酸堆積於體內,侵害人體的神經與造血系統。
異戊酸血症是一種胺基酸代謝異常疾病,致病原因為第15對染色體上的IVD基因突變,造成異戊醯輔酶A去氫酶發生缺陷,無法分解異戊酸,導致大量異戊酸堆積於體內,侵害人體的神經與造血系統。 瓜胺酸血症是一種尿素循環代謝異常疾病,人體在代謝蛋白質和氨基酸後會產生有毒的血氨,需要經由肝臟進行尿素循環反應,將有毒性的血氨轉換成較無毒性的尿素,再經由尿液排除至體外,疾病依致病機轉分為兩大類。
瓜胺酸血症是一種尿素循環代謝異常疾病,人體在代謝蛋白質和氨基酸後會產生有毒的血氨,需要經由肝臟進行尿素循環反應,將有毒性的血氨轉換成較無毒性的尿素,再經由尿液排除至體外,疾病依致病機轉分為兩大類。 血友病為一種先天性血液凝固異常的遺傳疾病,因為基因缺陷而造成血液中凝血因子的缺乏,導致身體的凝血機轉無法正常運作。
血友病為一種先天性血液凝固異常的遺傳疾病,因為基因缺陷而造成血液中凝血因子的缺乏,導致身體的凝血機轉無法正常運作。 天冬氨酰葡萄糖胺尿症致病原因為位於第4號染色體上的AGA基因突變,導致溶小體中缺乏天冬氨酰葡萄糖苷酶,進而阻斷醣蛋白的正常分解而大量堆積在溶小體內。
天冬氨酰葡萄糖胺尿症致病原因為位於第4號染色體上的AGA基因突變,導致溶小體中缺乏天冬氨酰葡萄糖苷酶,進而阻斷醣蛋白的正常分解而大量堆積在溶小體內。 低磷酸酯酶症是一種影響骨骼及牙齒發育的疾病,其致病原因為第1號染色體的ALPL基因發生突變,導致非特異性組織鹼性磷酸酶功能異常,無法參與骨骼組織礦化的過程,故無法形成堅硬的骨骼及牙齒。
低磷酸酯酶症是一種影響骨骼及牙齒發育的疾病,其致病原因為第1號染色體的ALPL基因發生突變,導致非特異性組織鹼性磷酸酶功能異常,無法參與骨骼組織礦化的過程,故無法形成堅硬的骨骼及牙齒。 神經節苷脂儲積症為一代謝性遺傳疾病,致病原因為第3號染色體上的GLB1基因突變,使得溶小體中β-galactosidase酵素功能不良,造成無法分解GM1神經節苷脂,進而堆積於組織及器官中,影響大腦和脊髓神經細胞。
神經節苷脂儲積症為一代謝性遺傳疾病,致病原因為第3號染色體上的GLB1基因突變,使得溶小體中β-galactosidase酵素功能不良,造成無法分解GM1神經節苷脂,進而堆積於組織及器官中,影響大腦和脊髓神經細胞。 戊二酸血症第2型是一種脂肪酸氧化過程異常疾病,其致病原因為ETFA、ETFB或ETFDH基因變異,造成體內無法分解脂肪、蛋白質以及膽鹼,使得血液和組織變得過於酸性,形成代謝性酸中毒。
戊二酸血症第2型是一種脂肪酸氧化過程異常疾病,其致病原因為ETFA、ETFB或ETFDH基因變異,造成體內無法分解脂肪、蛋白質以及膽鹼,使得血液和組織變得過於酸性,形成代謝性酸中毒。 戊二酸血症第1型是一種胺基酸代謝異常疾病,其致病原因為GCDH基因變異,使戊二基輔酶A去氫酵素異常,使得有毒的代謝中間產物戊二酸過量堆積,導致日後生理發育障礙與智能遲緩。
戊二酸血症第1型是一種胺基酸代謝異常疾病,其致病原因為GCDH基因變異,使戊二基輔酶A去氫酵素異常,使得有毒的代謝中間產物戊二酸過量堆積,導致日後生理發育障礙與智能遲緩。 甲狀腺是人體中重要的內分泌器官之一,主要維持體內新陳代謝功能,若發育中的幼童缺少甲狀腺素,會造成生長遲緩及智力不足。若當甲狀腺激素生成之相關基因發生變異時,會造成甲狀腺合成途徑出現問題,導致甲狀腺激素缺少而致病。
甲狀腺是人體中重要的內分泌器官之一,主要維持體內新陳代謝功能,若發育中的幼童缺少甲狀腺素,會造成生長遲緩及智力不足。若當甲狀腺激素生成之相關基因發生變異時,會造成甲狀腺合成途徑出現問題,導致甲狀腺激素缺少而致病。 多發性羧化酶缺乏症,其致病原因為HLCS基因突變,使全羧化酶合成酶缺乏,全羧化酶合成酶可以和生物素結合去幫助分解蛋白質、脂質及碳水化合物,一旦HLCS基因產生致病突變,就會降低此酵素結合生物素的能力,造成多種羧化酶功能異常,阻止營養物質的分解,破壞細胞功能,而造成症狀表現。
多發性羧化酶缺乏症,其致病原因為HLCS基因突變,使全羧化酶合成酶缺乏,全羧化酶合成酶可以和生物素結合去幫助分解蛋白質、脂質及碳水化合物,一旦HLCS基因產生致病突變,就會降低此酵素結合生物素的能力,造成多種羧化酶功能異常,阻止營養物質的分解,破壞細胞功能,而造成症狀表現。 丙酸血症為先天性代謝異常疾病,致病原因為PCCA或PCCB基因發生變異所致,使得丙醯基輔酵素A羧化酵素功能異常,導致大量堆積丙酸於體內發生代謝性酮酸中毒。
丙酸血症為先天性代謝異常疾病,致病原因為PCCA或PCCB基因發生變異所致,使得丙醯基輔酵素A羧化酵素功能異常,導致大量堆積丙酸於體內發生代謝性酮酸中毒。 體染色體隱性遺傳多囊性腎臟病,其致病原因為PKHD1基因突變,造成嬰兒或幼童的肝臟及腎臟出現囊泡與纖維化特徵,嚴重情況下需做血液透析或進行腎臟移植。
體染色體隱性遺傳多囊性腎臟病,其致病原因為PKHD1基因突變,造成嬰兒或幼童的肝臟及腎臟出現囊泡與纖維化特徵,嚴重情況下需做血液透析或進行腎臟移植。 楓糖尿症是特殊支鏈胺基酸代謝異常疾病,致病原因為BCKDHA、BCKDHB、DBT基因發生變異所致,這三個基因中任一個基因變異會降低支鏈甲型酮酸脫氫酵素,造成支鏈胺基酸大量堆積及產生毒性,不僅對腦細胞造成傷害,也會產生如楓樹糖漿的特殊體味。
楓糖尿症是特殊支鏈胺基酸代謝異常疾病,致病原因為BCKDHA、BCKDHB、DBT基因發生變異所致,這三個基因中任一個基因變異會降低支鏈甲型酮酸脫氫酵素,造成支鏈胺基酸大量堆積及產生毒性,不僅對腦細胞造成傷害,也會產生如楓樹糖漿的特殊體味。 范可尼氏貧血症為一種遺傳性骨髓造血功能異常的疾病,其相關基因與DNA修復有關,當基因發生缺陷時會導致造血幹細胞異常凋亡,無法分化為成熟的血球細胞,造成骨髓造血功能失常,另外患者可能會有急性白血病或其他癌症等症狀。
范可尼氏貧血症為一種遺傳性骨髓造血功能異常的疾病,其相關基因與DNA修復有關,當基因發生缺陷時會導致造血幹細胞異常凋亡,無法分化為成熟的血球細胞,造成骨髓造血功能失常,另外患者可能會有急性白血病或其他癌症等症狀。 高雪氏症是一種溶小體神經脂質儲積症,致病原因為第1號染色體上的GBA基因突變,使得葡萄糖腦甘脂酵素活性降低,造成代謝物異常堆積在肝、骨髓、脾臟及腦部等處,進而導致器官損傷或智力減退等症狀。
高雪氏症是一種溶小體神經脂質儲積症,致病原因為第1號染色體上的GBA基因突變,使得葡萄糖腦甘脂酵素活性降低,造成代謝物異常堆積在肝、骨髓、脾臟及腦部等處,進而導致器官損傷或智力減退等症狀。 裘馨氏/貝克氏肌肉萎縮症是一種進行性肌肉無力及萎縮之遺傳性疾病,其致病原因為X染色體上DMD基因變異,造成肌縮蛋白生成異常,影響肌肉細胞的完整性,導致肌肉細胞逐漸萎縮及壞死。
裘馨氏/貝克氏肌肉萎縮症是一種進行性肌肉無力及萎縮之遺傳性疾病,其致病原因為X染色體上DMD基因變異,造成肌縮蛋白生成異常,影響肌肉細胞的完整性,導致肌肉細胞逐漸萎縮及壞死。 苯酮尿症是一種代謝異常的遺傳性疾病,因致病機轉不同,可分為兩大型別,PAH和BH4缺乏型,因苯丙胺酸堆積無法正常代謝,會造成神經系統傷害,若無早期發現及治療,患者會有發育不良及智能障礙等症狀。
苯酮尿症是一種代謝異常的遺傳性疾病,因致病機轉不同,可分為兩大型別,PAH和BH4缺乏型,因苯丙胺酸堆積無法正常代謝,會造成神經系統傷害,若無早期發現及治療,患者會有發育不良及智能障礙等症狀。 瓦登伯革氏症候群又被稱為藍眼珠症候群,患者的眼珠虹膜異色,但視力並未受到影響,易合併聽障及長期便秘等問題。
瓦登伯革氏症候群又被稱為藍眼珠症候群,患者的眼珠虹膜異色,但視力並未受到影響,易合併聽障及長期便秘等問題。 遺傳性球形紅血球增多症是由調控紅血球細胞膜蛋白基因異常所引起的遺傳性疾病,此疾病兩大特徵為紅血球不足及脾臟中過多的紅血球。
遺傳性球形紅血球增多症是由調控紅血球細胞膜蛋白基因異常所引起的遺傳性疾病,此疾病兩大特徵為紅血球不足及脾臟中過多的紅血球。 第2型膠原蛋白又稱軟骨膠原蛋白,主要參與軟骨細胞合成,主要存在的區域為軟骨組織、眼球玻璃體內、內耳及脊椎骨之間的椎間盤中心(髓核)等部位。當COL2A1基因發生變異時,就會造成軟骨發育不良,而導致臨床症狀出現。
第2型膠原蛋白又稱軟骨膠原蛋白,主要參與軟骨細胞合成,主要存在的區域為軟骨組織、眼球玻璃體內、內耳及脊椎骨之間的椎間盤中心(髓核)等部位。當COL2A1基因發生變異時,就會造成軟骨發育不良,而導致臨床症狀出現。 Nonaka肌病變致病原因為GNE基因發生變異所致,使得細胞表面的唾液酸降低,造成細胞移動、附著及訊號傳遞等受到影響,導致漸進性的遠端肌無力。
Nonaka肌病變致病原因為GNE基因發生變異所致,使得細胞表面的唾液酸降低,造成細胞移動、附著及訊號傳遞等受到影響,導致漸進性的遠端肌無力。 球細胞腦白質失養症其致病原因為GALC基因變異所致,使得半乳糖腦苷酯脢酵素活性缺乏無法正常代謝,進而大量堆積在神經細胞內,引發神經學特徵與症狀。
球細胞腦白質失養症其致病原因為GALC基因變異所致,使得半乳糖腦苷酯脢酵素活性缺乏無法正常代謝,進而大量堆積在神經細胞內,引發神經學特徵與症狀。 史黛氏症是一種罕見的遺傳性疾病,其致病原因是SBDS基因發生變異,使得參與製造蛋白質的核醣體異常,會影響骨髓、胰臟及骨骼等器官發育。
史黛氏症是一種罕見的遺傳性疾病,其致病原因是SBDS基因發生變異,使得參與製造蛋白質的核醣體異常,會影響骨髓、胰臟及骨骼等器官發育。 原發性肉鹼缺乏症為一種脂肪酸氧化異常疾病,患者因體內肉鹼缺乏,使得無法有效利用脂肪酸來產生能量,也無法產生足夠的酮體為大腦使用,導致可能出現心肌、腦神經、肌肉骨骼等方面出現病變。
原發性肉鹼缺乏症為一種脂肪酸氧化異常疾病,患者因體內肉鹼缺乏,使得無法有效利用脂肪酸來產生能量,也無法產生足夠的酮體為大腦使用,導致可能出現心肌、腦神經、肌肉骨骼等方面出現病變。 肝醣儲積症第1型是一種遺傳代謝疾病,使患者缺乏葡萄糖-6-磷酸酵素功能,造成肝醣無法順利轉化成葡萄糖,除了發生低血糖外,也會導致體內累積過多肝醣,長期下來會損害多重臟器功能。
肝醣儲積症第1型是一種遺傳代謝疾病,使患者缺乏葡萄糖-6-磷酸酵素功能,造成肝醣無法順利轉化成葡萄糖,除了發生低血糖外,也會導致體內累積過多肝醣,長期下來會損害多重臟器功能。 半乳糖血症是一種醣類代謝異常的疾病,使得半乳糖轉變成葡萄糖的代謝途徑發生障礙,導致體內半乳糖的堆積,造成生長及發展遲緩、智力障礙、肝臟疾病及腎臟問題等。
半乳糖血症是一種醣類代謝異常的疾病,使得半乳糖轉變成葡萄糖的代謝途徑發生障礙,導致體內半乳糖的堆積,造成生長及發展遲緩、智力障礙、肝臟疾病及腎臟問題等。 囊腫纖維症其致病原因為CFTR 基因突變所致,導致患者的呼吸道、消化道、胰臟及汗腺等外分泌腺體器官功能異常,造成分泌物無法順利排出體外,增加感染風險。
囊腫纖維症其致病原因為CFTR 基因突變所致,導致患者的呼吸道、消化道、胰臟及汗腺等外分泌腺體器官功能異常,造成分泌物無法順利排出體外,增加感染風險。 蠶豆症是一種很常見的先天性代謝異常疾病,因G6PD基因變異會導致紅血球內G6PD酵素活性減少,當碰到特定氧化物質時,易產生溶血、貧血等症狀。
蠶豆症是一種很常見的先天性代謝異常疾病,因G6PD基因變異會導致紅血球內G6PD酵素活性減少,當碰到特定氧化物質時,易產生溶血、貧血等症狀。 家族性地中海熱其致病原因為MEFV基因突變所致,造成患者白血球中的比林蛋白活性降低,使得體內發炎反應無法正常調節,患者會反覆發燒、胸腹部和關節處疼痛等臨床症狀。
家族性地中海熱其致病原因為MEFV基因突變所致,造成患者白血球中的比林蛋白活性降低,使得體內發炎反應無法正常調節,患者會反覆發燒、胸腹部和關節處疼痛等臨床症狀。 血鐵沉積症,又稱「血色素沉著病」是一種小腸黏膜對鐵吸收過多鐵的疾病。臨床特徵是肝臟、皮膚、胰腺、心臟、關節和垂體前葉過度儲存鐵,在未經治療的患者中,早期症狀包括:腹痛、虛弱、嗜睡、體重減輕、關節痛等。長期鐵的累積會造成身體臟器受損。
血鐵沉積症,又稱「血色素沉著病」是一種小腸黏膜對鐵吸收過多鐵的疾病。臨床特徵是肝臟、皮膚、胰腺、心臟、關節和垂體前葉過度儲存鐵,在未經治療的患者中,早期症狀包括:腹痛、虛弱、嗜睡、體重減輕、關節痛等。長期鐵的累積會造成身體臟器受損。 威爾森氏症是一種罕見的遺傳疾病,患者時常因為被忽略病因而延誤了診斷與治療,發病年齡多為六至四十歲;其臨床症狀和發病年齡有個體差異性,從孩童時期到青壯年都有可能發病,疾病嚴重度也有很大的差異。
威爾森氏症是一種罕見的遺傳疾病,患者時常因為被忽略病因而延誤了診斷與治療,發病年齡多為六至四十歲;其臨床症狀和發病年齡有個體差異性,從孩童時期到青壯年都有可能發病,疾病嚴重度也有很大的差異。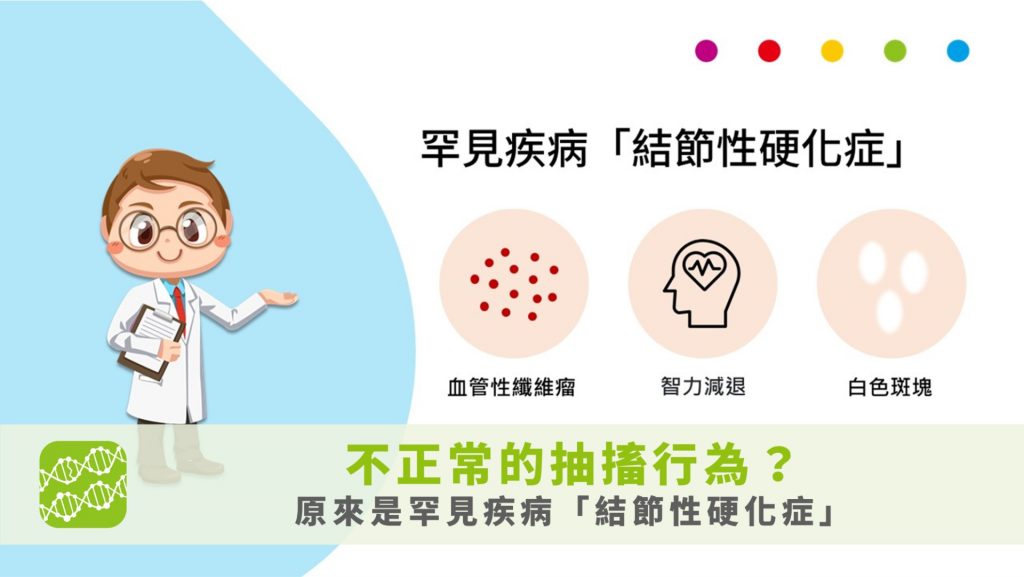 5歲大的奇奇看起來活潑健康,但就在某個假日,奇奇的爺爺帶著到公園玩的時候,玩著玩著爺爺發現奇奇突然出現不正常的抽搐行為…
5歲大的奇奇看起來活潑健康,但就在某個假日,奇奇的爺爺帶著到公園玩的時候,玩著玩著爺爺發現奇奇突然出現不正常的抽搐行為… 患者常常給人的第一印象就是高高瘦瘦的且手腳也長,因為有這樣的特徵,常會提拔為運動校隊或選手,若在不知情的狀態下進行激烈運動或訓練時,往往會造成心臟負擔,嚴重時會猝死危及生命安全
患者常常給人的第一印象就是高高瘦瘦的且手腳也長,因為有這樣的特徵,常會提拔為運動校隊或選手,若在不知情的狀態下進行激烈運動或訓練時,往往會造成心臟負擔,嚴重時會猝死危及生命安全 肝醣儲積症第二型(Glycogen Storage Disease type II)又稱龐貝氏症(Pompe Disease);患者最主要臨床症狀為肌肉相關問題,以肌肉張力降低、無力為主。龐貝氏症依照發病時間點可分為嬰兒型和晚發型兩種。
肝醣儲積症第二型(Glycogen Storage Disease type II)又稱龐貝氏症(Pompe Disease);患者最主要臨床症狀為肌肉相關問題,以肌肉張力降低、無力為主。龐貝氏症依照發病時間點可分為嬰兒型和晚發型兩種。 依據中華民國兒童癌症基金會統計,「視網膜母細胞瘤」為台灣十大兒童癌症之一,亦是兒童最常見的眼科癌症。
依據中華民國兒童癌症基金會統計,「視網膜母細胞瘤」為台灣十大兒童癌症之一,亦是兒童最常見的眼科癌症。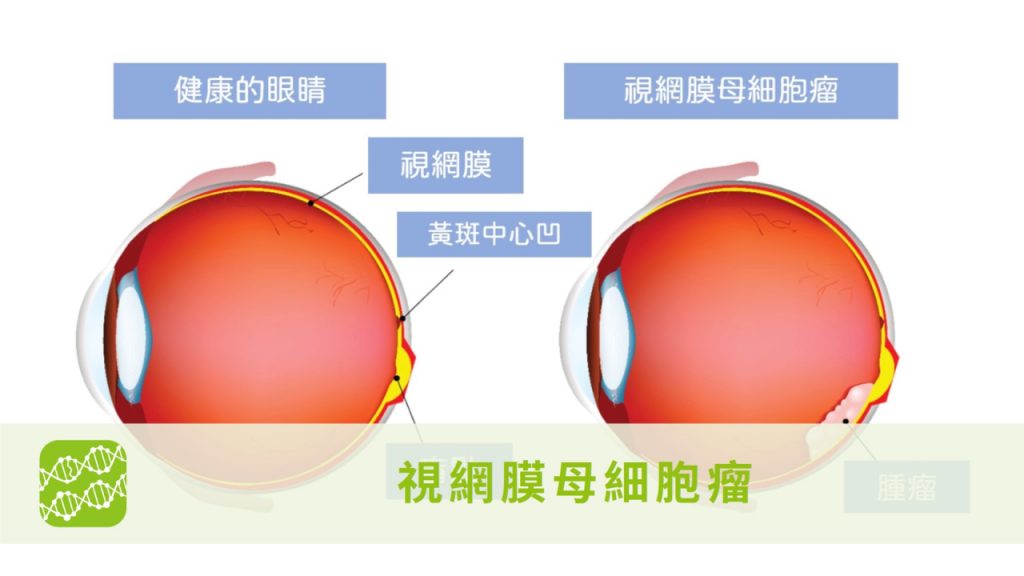 為一種較為罕見的眼部腫瘤疾病,是在兒童眼睛腫瘤中最常見的原發性惡性腫瘤,大部份發病的年齡介於0~5歲的嬰幼兒中
為一種較為罕見的眼部腫瘤疾病,是在兒童眼睛腫瘤中最常見的原發性惡性腫瘤,大部份發病的年齡介於0~5歲的嬰幼兒中 眼睛皮膚白化症是一種影響皮膚、頭髮和眼睛著色(色素沉著)的遺傳疾病,其致病原因為相關基因變異導致所製造的蛋白有缺陷,使得色素細胞製造出的黑色素減少
眼睛皮膚白化症是一種影響皮膚、頭髮和眼睛著色(色素沉著)的遺傳疾病,其致病原因為相關基因變異導致所製造的蛋白有缺陷,使得色素細胞製造出的黑色素減少 造成身材矮小的原因,除了年齡、營養、壓力、生活型態之外,研究指出SHOX(矮小基因)基因也會導致嚴重影響
造成身材矮小的原因,除了年齡、營養、壓力、生活型態之外,研究指出SHOX(矮小基因)基因也會導致嚴重影響 發病時間從兒童時期至成人時期皆有可能,患者在臨床症狀上通常為對稱性的,從手部和腿部的遠端運動神經開始病變,而導致手掌與腳掌的肌肉逐漸無力與開始萎縮,讓患者在行動與生活造成困難與不便
發病時間從兒童時期至成人時期皆有可能,患者在臨床症狀上通常為對稱性的,從手部和腿部的遠端運動神經開始病變,而導致手掌與腳掌的肌肉逐漸無力與開始萎縮,讓患者在行動與生活造成困難與不便 是由於類固醇-21 羥化脢(Steroid 21-hydroxylase, CYP21A2)的基因變異所造成,因此基因變異導致先天缺少某些製造腎上腺皮質素的酵素,進而造成腎上腺疾病;依酵素缺乏程度可能有程度不一的男性化徵象,嚴重者會有鈉和水分不平衡等問題
是由於類固醇-21 羥化脢(Steroid 21-hydroxylase, CYP21A2)的基因變異所造成,因此基因變異導致先天缺少某些製造腎上腺皮質素的酵素,進而造成腎上腺疾病;依酵素缺乏程度可能有程度不一的男性化徵象,嚴重者會有鈉和水分不平衡等問題 先天性腎上腺增生症的酵素數值異常,在臨床上嚴重者可能會有嗜睡嘔吐、或是性器官混淆的狀況,輕微者可能會有性早熟情況發生
先天性腎上腺增生症的酵素數值異常,在臨床上嚴重者可能會有嗜睡嘔吐、或是性器官混淆的狀況,輕微者可能會有性早熟情況發生 是肌肉萎縮症中最常見的一種,根據歐美統計每十萬新生男嬰中有約20~30人患病,其致病原因是位於X染色體短臂Xp21的Dystrophin基因(DMD gene)有異常而造成的
是肌肉萎縮症中最常見的一種,根據歐美統計每十萬新生男嬰中有約20~30人患病,其致病原因是位於X染色體短臂Xp21的Dystrophin基因(DMD gene)有異常而造成的 粒線體基因突變大部份為遺傳性,若當粒線體缺陷時,就無法釋出足夠的能量,對於需要大量能量的器官就會產生病變
粒線體基因突變大部份為遺傳性,若當粒線體缺陷時,就無法釋出足夠的能量,對於需要大量能量的器官就會產生病變 蛋白質C缺乏症(Protein C deficiency)是一種會讓血液異常凝結風險增加的疾病,其致病原因為PROC基因變異使得製造蛋白質C的功能受影響而導致
蛋白質C缺乏症(Protein C deficiency)是一種會讓血液異常凝結風險增加的疾病,其致病原因為PROC基因變異使得製造蛋白質C的功能受影響而導致 為什麼小華看起來好好的,卻有這個基因異常?在網路上查到關於蛋白質C缺乏症患者的臨床症狀,未來是不是都會出現在小華身上?
為什麼小華看起來好好的,卻有這個基因異常?在網路上查到關於蛋白質C缺乏症患者的臨床症狀,未來是不是都會出現在小華身上? 目前美國FDA核准三種藥物可治療脊髓性肌肉萎縮症,可以改善患者的肌肉運動功能並且提升整體存活率
目前美國FDA核准三種藥物可治療脊髓性肌肉萎縮症,可以改善患者的肌肉運動功能並且提升整體存活率 李爸爸、李媽媽發現每當帶著倫倫站立的時候,倫倫的腳好像都沒什麼力氣。透過臨床診斷以及基因檢查,確定了倫倫罹患了脊髓性肌肉萎縮症
李爸爸、李媽媽發現每當帶著倫倫站立的時候,倫倫的腳好像都沒什麼力氣。透過臨床診斷以及基因檢查,確定了倫倫罹患了脊髓性肌肉萎縮症 脊髓性肌肉萎縮症(Spinal Muscular Atrophy, SMA)是一種可能致命的遺傳性疾病。發病年齡從出生到成年皆有可能發生,每個人依照發病年齡以及疾病的嚴重程度,症狀會有很大的差異
脊髓性肌肉萎縮症(Spinal Muscular Atrophy, SMA)是一種可能致命的遺傳性疾病。發病年齡從出生到成年皆有可能發生,每個人依照發病年齡以及疾病的嚴重程度,症狀會有很大的差異 目前有越來越多的疾病被發現跟基因有關係。基因檢測在目前科學的應用有哪些呢?主要可以分為疾病診斷、預防醫學、精準醫療三個方面
目前有越來越多的疾病被發現跟基因有關係。基因檢測在目前科學的應用有哪些呢?主要可以分為疾病診斷、預防醫學、精準醫療三個方面 關於家族性澱粉樣多發性神經病變之標靶治療用藥分析
關於家族性澱粉樣多發性神經病變之標靶治療用藥分析 是一種罕見疾病,依不同的疾病類型與種族,其發生率與發病年齡也有所不同。臨床特徵可包括周圍感覺運動神經病變和自主神經病變
是一種罕見疾病,依不同的疾病類型與種族,其發生率與發病年齡也有所不同。臨床特徵可包括周圍感覺運動神經病變和自主神經病變 肌肉細胞中某部分的蛋白質無法獲得適當營養,進而出現變性壞死或造成結構變化,因此出現肌肉力量降低的症狀
肌肉細胞中某部分的蛋白質無法獲得適當營養,進而出現變性壞死或造成結構變化,因此出現肌肉力量降低的症狀 此疾病與血管生成及修補異常有關,非血液凝固問題或凝血因子缺乏所造成。患者通常有反覆性鼻出血的症狀,平均發病年齡為12歲
此疾病與血管生成及修補異常有關,非血液凝固問題或凝血因子缺乏所造成。患者通常有反覆性鼻出血的症狀,平均發病年齡為12歲 芯芯媽媽想起自己舅舅經常性的流鼻血,但自己並沒有這樣的情況,有可能會是自己遺傳給女兒嗎?
芯芯媽媽想起自己舅舅經常性的流鼻血,但自己並沒有這樣的情況,有可能會是自己遺傳給女兒嗎?