畫作名稱:圖書館員的一天
畫作介紹:我未來的職業夢想是當圖書館管理員,可以閱讀好多好多書,透過書籍豐富自己的視野。
作者名稱:蔡永濬
個案故事
我是罕見疾病-裘馨氏肌肉失養症的患者,從寶寶時期學走路就比其他小朋友慢,也連帶著學習遲緩一些。 慶幸家人發現得早,讓我從小學游泳、參加醫院的早療學習….
由於我是基因點缺損,相對同病症的患者,我的狀況算是很棒的,24歲還有行的能力! 只是無法走太久,或做劇烈的活動。
慶幸自己生在這個時代,日常的活動,都還有電動輪椅的輔助! 它帶著我為自己的旅程開啟另一扇窗。
疾病名稱
裘馨氏肌肉失養症
疾病介紹
裘馨氏肌肉萎縮症是一種進行性肌肉無力及萎縮的遺傳性疾病,其致病原因是位於X染色體上DMD基因變異,造成肌縮蛋白(dystrophin)生成異常,影響肌肉細胞的完整性,導致肌肉細胞逐漸萎縮及壞死。有此基因異常者會導致本身肌肉組織隨著年紀衰退外,於晚期也可能因控制呼吸與行動的肌肉萎縮,而導致其他併發症甚至有死亡的風險。
臨床症狀
男性患者剛出生時發育大致正常且無明顯症狀,隨著年紀增長肌肉組織會逐漸衰退,通常在5歲前即發病,患者的肌肉組織逐漸由脂肪和纖維組織取代,首先下肢的肌肉會有漸進性衰退現象,出現走路經常跌倒、上下樓梯較為吃力等症狀。患者約10幾歲開始因無法走動或站立需要倚靠輪椅行動,漸漸因呼吸道肌肉功能喪失而需要呼吸維持器;另外多數患者會於18歲前發生心肌病變造成心臟衰竭。呼吸系統併發症(如肺部感染、呼吸衰竭)和心肌病變為常見造成患者死亡的原因,患者壽命約30歲左右。女性帶因者通常無症狀或有輕微肌肉衰退等症狀。
遺傳模式
性聯遺傳。女性若帶有一個DMD基因突變點位,應為帶因者,但由於X染色體去活化機制,亦有可能出現輕微的症狀;男性則因僅有一條X染色體,故只要帶有此基因突變點位,即為患者。
檢驗方式
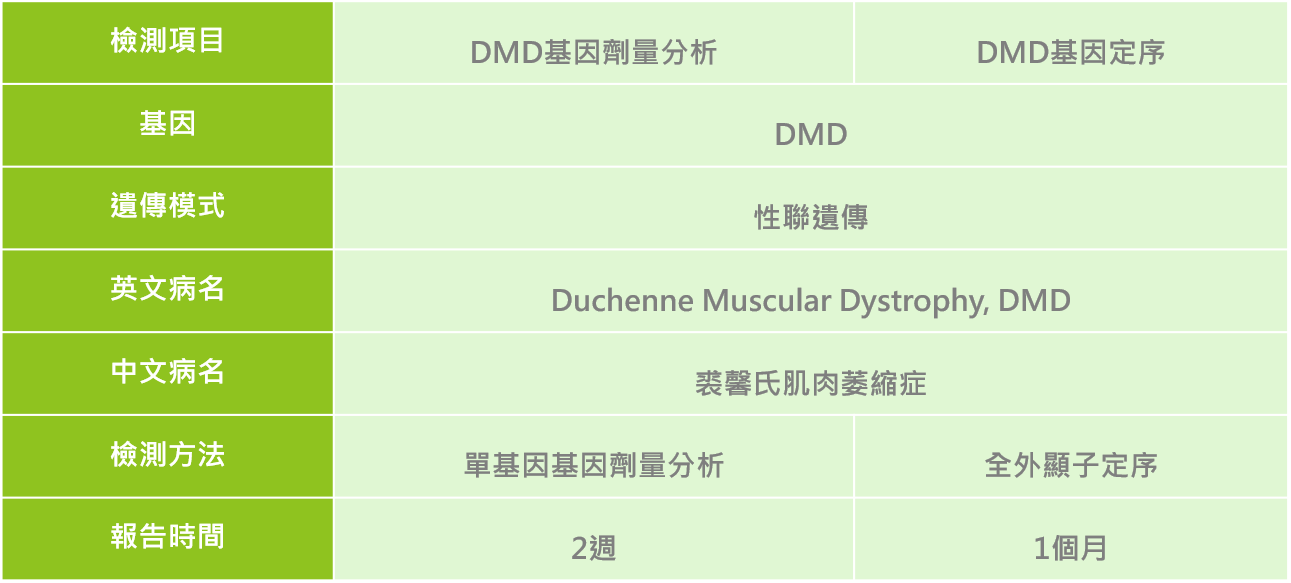
目前可透過專業醫師評估後進行酵素檢測、肌電圖、肌肉切片及心電圖等影像學來做為疾病診斷,此外也可以利用分子檢測方式進行DMD基因檢測;針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓裘馨氏肌肉萎縮症患者可以及早診斷治療與醫療協助。
參考資料
- 財團法人罕見疾病基金會
- GeneReviews®
- 罕見遺傳疾病一點通
- 中華民國肌萎縮症病友協會