

疾病簡介
球細胞腦白質失養症(Globoid Cell Leukodystrophy)又稱Krabbe氏症(Krabbe disease),致病原因為位於第14號染色體上的GALC基因突變所致,導致半乳糖腦苷酯脢(GALC)酵素活性缺乏,使半乳糖腦苷酯及鞘氨醇半乳糖苷無法正常被分解,大量堆積在製造髓鞘的神經細胞中,且鞘氨醇半乳糖苷的毒性會破壞製造髓鞘的細胞,造成包覆在神經細胞上的髓鞘逐漸脫落消失,而失去髓鞘的神經細胞將無法正常執行功能,引發Krabbe氏症的神經學特徵與症狀。GALC基因帶因率約為0.6%。
臨床症狀
此疾病可區分為嬰兒型和晚發型,嬰兒型通常於出生6個月內發病,平均壽命為13個月左右,臨床症狀有焦躁不安、肌肉張力過高、餵食困難、漸進性神經功能退化、非感染性發燒、視力喪失、癲癇及身心發展遲緩且退化等;晚發型的患者其臨床症狀有個體差異性,通常於出生6個月後的任意時間點發病,而部分患者於青春期或成年後才發病,會出現肌肉無力、視力障礙、智力減退等臨床症狀。治療上主要以支持性治療為主,避免併發症發生,發病的早期可考慮進行造血幹細胞移植,可維持部分的神經功能。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有 1/4 的機率生下患者。
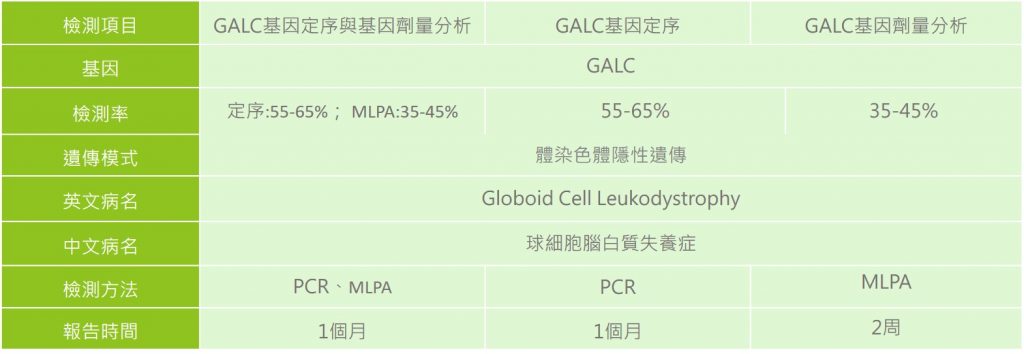
檢驗方式
患者會進行白血球或皮膚纖維母細胞的GALC酵素活性分析、神經學檢查、腦電圖檢查(EGG)以及腦部磁振造影檢查(MRI);另外,透過基因分子檢測技術也可以提供進一步的診斷。
首選慧智(相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師
參考資料
- J Inherit Metab Dis. 1999;22(2):155-62.
- GeneReviews®
- National Institutes of Health
- 罕見疾病一點通



