
疾病簡介
先天性腎上腺增生症(Congenital adrenal hyperplasia, CAH)是由於類固醇-21 羥化脢(Steroid 21-hydroxylase, CYP21A2)的基因變異所造成,因此基因變異導致先天缺少某些製造腎上腺皮質素的酵素,進而造成腎上腺疾病;依酵素缺乏程度可能有程度不一的男性化徵象,嚴重者會有鈉和水分不平衡等問題。
腎上腺皮質所分泌的類固醇中最主要的有鹽皮素(mineralocorticoid)、糖皮素(glucocorticoid)及雄性素(androgen)等三種。分別作用為:鹽皮素促成鹽分的積留於體內,糖皮素則與葡萄糖代謝及人體應付環境壓力的反應有關,雄性素則與人類雄性化有關。
臨床症狀
先天性腎上腺增生症(Congenital adrenal hyperplasia, CAH) ,依症狀的嚴重度及出現的遲早,可分典型與非典型兩大類型:
1. 典型-失鹽型:
新生兒期即會因鹽分的大量流失造成低血鈉,於出生不久會出現嗜睡嘔吐、生長遲滯、電解質流失、高血鉀;若未積極給予治療,嚴重時則可能導致急性腎上腺機能不全引發休克、猝死。失鹽型患者需終生服用皮質素和留鹽激素,並於飲食中添加適量的鹽分,用以補充流失的鈉離子。
2. 典型-單純男性化型:
由於體內雄性素大量分泌的作用下,使得女嬰在胎兒期發生外陰部雄性化,出生時常會有性器混淆或陰蒂過長現象;而男嬰的外生殖器正常較難以早期診斷,直到孩童時期則會出現性早熟現象時才可能會被發現。單純男性化型患者需終生補充皮質素。
3. 非典型-晚發型:
多數於兒童期或青春期出現雄性素過高,如青春痘、陰毛過早生長、生長過速、骨齡提前,女性患者在出生時具有正常生殖器,青春期時可能會出現多毛、初經延遲、月經不規則等。男性患者通常無症狀,但可能出現鬍鬚過早出現、陰莖增大等。晚發型女性患者在治療方面使用低劑量的皮質素,若程度嚴重者則需要整型手術矯正,男性患者若沒有嚴重症狀則不需要治療。
遺傳模式
先天性腎上腺增生症(Congenital adrenal hyperplasia, CAH)遺傳模式為體染色體隱性遺傳,通常夫妻雙方為此疾病之帶因者,則下一代有1/4的機率生下患病之胎兒,3/4的機率生下正常之胎兒。
根據國外文獻資料發生率約為1/15000,而在台灣,依據新生兒篩檢中心的資料所顯示之發生率約為1/10000-15000。
檢驗方式
先天性腎上腺增生症(Congenital adrenal hyperplasia, CAH)現在可透過新生兒篩檢的方式,測定血片中17-OHP的含量為篩檢的指標;若濃度高於正常值,將接獲通知並依照指示回診。先天性腎上腺增生症患者除臨床評估外,也應作血液染色體檢查,血中pH值、鉀、鈉 離子濃度及相關之皮質類固醇含量,尿液中則應檢查孕三醇(Pregnanetriol)及17-酮類固醇(17-ketosteroid)濃度協助疾病診斷。
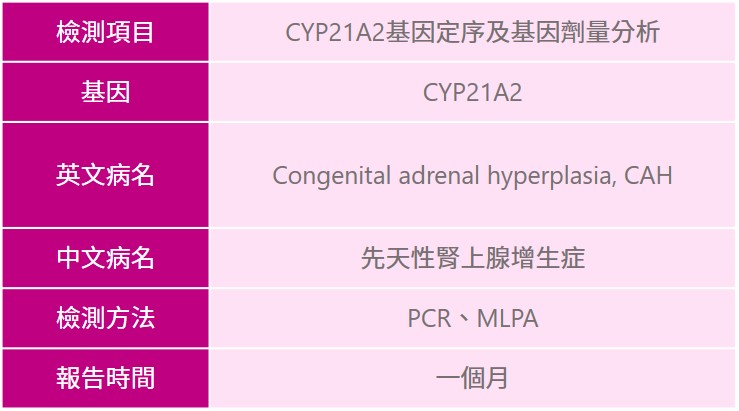
針對最常見的CYP21A2基因,也可以透過醫師臨床評估後用分子檢測方式進行基因檢測,有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢;確診的患者透過評估可進行手術治療外,如果能及時治療並持續追蹤,此疾病的預後是較為良好的。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
● GeneReviews®
● 財團法人罕見疾病基金會
● 罕見遺傳疾病一點通
● 財團法人中華民國衛生保健基金會 新生兒篩檢中心



