

疾病簡介
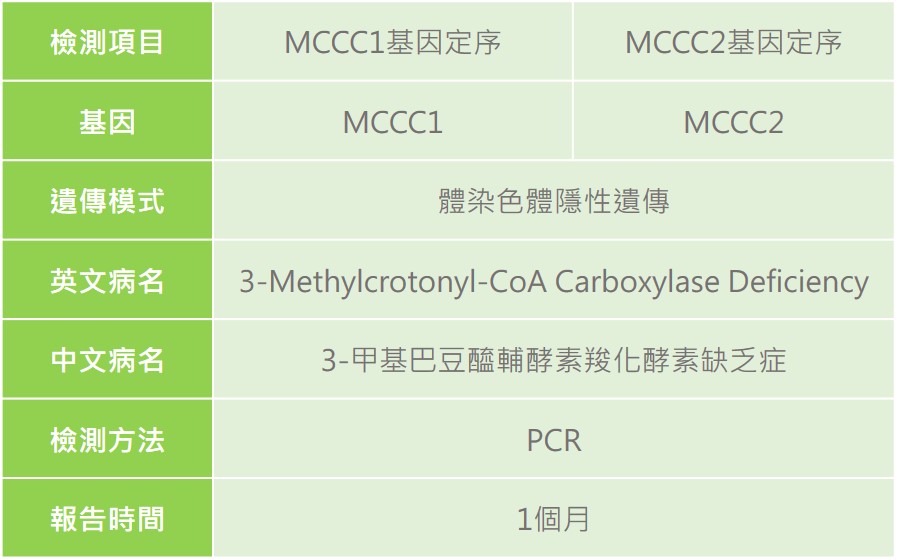
3-甲基巴豆醯輔酵素羧化酵素缺乏症(3-Methylcrotonyl-CoA Carboxylase Deficiency, 3-MCC Deficiency)是一種代謝異常疾病。因致病基因不同分為2種型別,第1型致病原因為第3對染色體上的MCCC1或稱MCCA (Methylcrotonyl-CoA carboxylase alpha chain)基因突變,第2型致病原因為第5對染色體上的MCCC2或稱MCCB (Methylcrotonyl-CoA carboxylase beta chain)基因突變。這兩個基因發生突變會導致3-甲基巴豆醯輔酶酵素羧化酵素活性降低,無法有效代謝白胺酸,造成三甲基巴豆醯輔酶甘胺酸(3-methylcrotonylglycine)逐漸累積於體內造成毒性,對人體造成影響。
MCCC1及MCCC2基因帶因率在台灣地區約為1.9%。
臨床症狀
患者通常出生時並無症狀,於嬰兒期或兒童早期受到感染、發燒或長時間飢餓才出現臨床症狀,臨床症狀個體差異較大,可能出現餵食困難、持續性嘔吐、腹瀉、脫水以及嗜睡等症狀,過多白胺酸累積於體內會導致代謝性酸中毒、高血氨、低血糖、肌張力低下等現象,如果出現症狀無妥善治療,會造成嚴重代謝機能衰退或生長發育遲緩,嚴重時可能導致昏迷或死亡。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,但下一代不分性別有1/4的機率為患者。
檢驗方式
目前可透過新生兒篩檢檢測體內3-羥基異戊醯肉鹼(3-Hydroxyisovaleryl Carnitine, C5OH)的濃度,當C5OH濃度較高,會建議進一步進行複檢及轉介。透過專業醫師的臨床評估,檢測患者血糖數值、血液酸鹼值、血氨、肝功能檢測、尿液等常規檢查,必要時可測定白血球、皮膚纖維母細胞中酵素活性診斷,或是進行MCCC1或MCCC2基因診斷。
針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓3-甲基巴豆醯輔酵素羧化酵素缺乏症之患者可以及早診斷治療與醫療協助。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- J Clin Invest. 2001 ;107(4):495-504.
- Orphanet J Rare Dis. 2012; 7: 31.
- National Institutes of Health
- 罕見疾病一點通



