

疾病簡介
阿拉吉歐症候群(Alagille syndrome)的患者90%是因JAG1基因突變所致,7%是第20號染色體短臂20p11.23位置發生缺失所致,此片段包含JAG1基因,另外僅1%的患者與NOTCH2基因的突變有關。
JAG1是位於細胞表面的蛋白質,可與NOTCH1的受體結合,若JAG1基因異常會造成NOTCH訊息傳遞異常,進而影響胚胎發育,如:胎兒器官的形成,像肝臟、心臟、腎臟、臉部外觀、骨骼以及眼睛等。此疾病於臨床表現具有個體差異性,同一家族中具有此變異之不同個體,可能會有程度不同的影響。
阿拉吉歐症候群的發生率約為1/70,000,因部分患者於嬰兒期不會發生肝臟疾病,因此可能低估疾病發生率。
臨床症狀
新生兒的生理性黃疸,通常在出生後1~2週就會自然痊癒,如果黃疸持續超過兩週以上,尤其滿月後黃疸仍未消退,便是所謂的「延遲性黃疸」,可能是新生兒的肝膽系統出了問題,而阿拉吉歐症候群也是造成新生兒延遲性黃疸的疾病之一。
臨床上患者除了有黃疸,還可能有先天性心臟血管疾病(周邊肺動脈狹窄、法洛氏四重症或更複雜的心臟疾病)、面部特徵(前額突出、眼距寬、眼窩深、下巴尖、鼻樑凸)、骨骼結構異常(蝴蝶狀脊椎骨),以及眼球角膜周邊出現渾濁的後胚胎環(Posterior embryotoxon)。
另外,患者因膽汁無法順利排出,其腸道內的膽鹽濃度變低,會影響到腸道脂肪的吸收,進而造成必需脂肪酸及脂溶性維生素的吸收不足,因此會有凝血功能異常(維生素K缺乏)、佝僂病(維生素D缺乏)、視網膜病變(維生素A和E缺乏)、周邊神經病變及肌肉病變(維生素E缺乏)。
患者因長期脂肪的吸收不良而造成身體熱量攝取不足,50~90%的患者會有生長遲緩的問題,且無法施打生長激素來改善。
遺傳模式
體染色體顯性遺傳,約30~50%的患者是遺傳自父母,若父母親其中一方帶有此片段缺失,則下一代有50%機率會遺傳到此疾病。部分患者為自發性變異(非父母遺傳造成),是由於生殖細胞(卵子或精子)形成過程或胎兒早期發育過程中,隨機發生染色體缺失或JAG1基因突變引起的。
檢驗方式
患者最常見的特徵為膽管缺少,因此可由檢測患者肝臟組織切片中膽管的數量作為診斷的考量,除了膽管缺少的情形外,也可藉由患者可能出現之臨床症狀(如:特殊的外觀長相、心臟疾病、骨骼異常等)作為診斷依據。
由於患者所表現出來的臨床表徵差異很大,因此阿拉吉歐症候群疾病診斷較不易,若疑似患者的一等親有罹患此症,建議一等親的臨床症狀都應納入診斷考量。
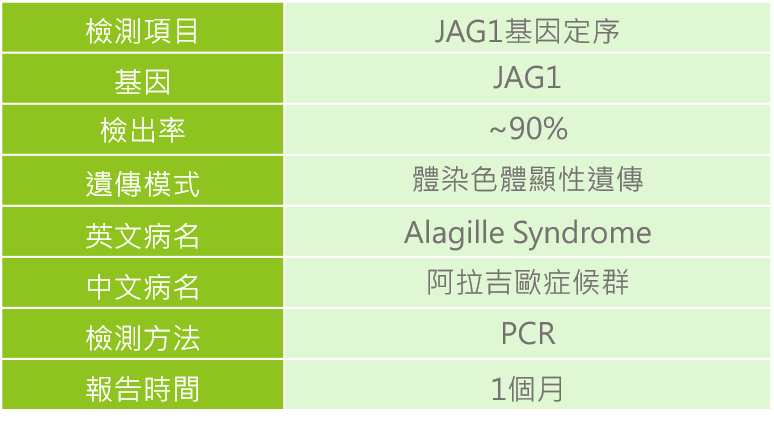
1. 針對有臨床特徵的患者或有家族疾病史的家族成員,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓阿拉吉歐症候群的患者可以及早診斷治療與醫療協助。由於90%阿拉吉歐症候群的患者是因JAG1基因突變所致,因此可透過分子基因檢測來診斷。
2. 確認胎兒是否為阿拉吉歐症候群,於產前可選擇進行染色體相關檢測,如非侵染色體篩檢(NIPS)、侵入性檢測-以絨毛膜或羊水檢體進行全方位複合式晶片檢查,確認胎兒是否為阿拉吉歐症候群,檢測前需與您的主治醫師進行評估與討論。
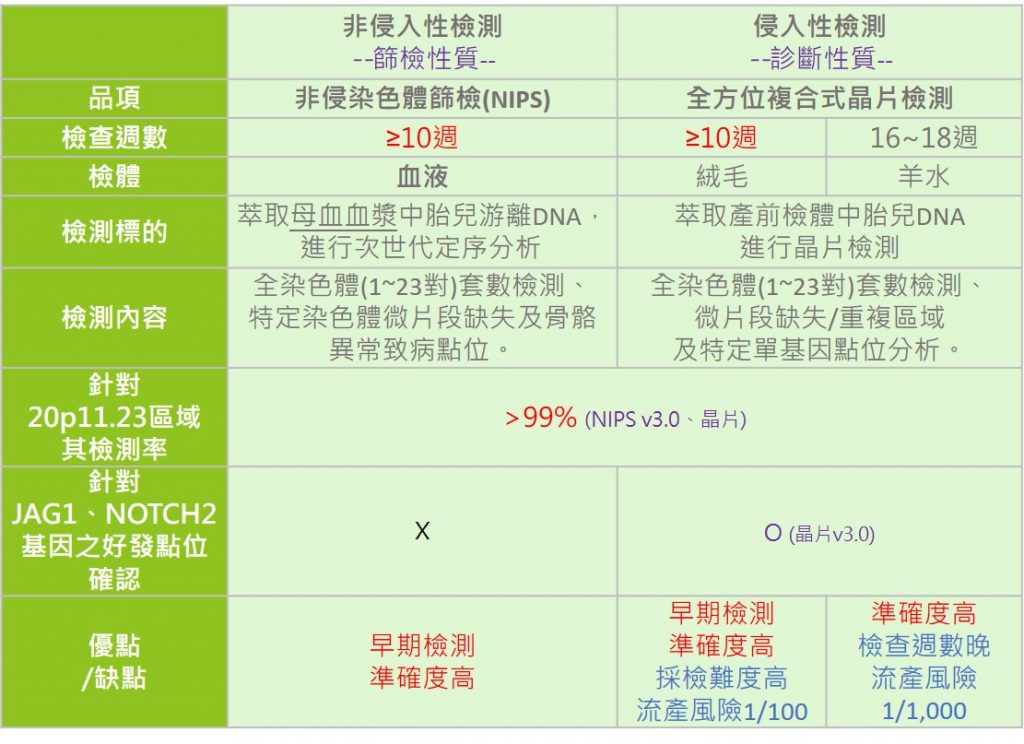
(註)部分疾病可能具有多種致病機轉,故檢測有其侷限性,無法呈現所有可能情況,若有需要進行特定基因檢測,請洽詢專業醫療人員或醫師。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- 罕見疾病一點通
- National Library of Medicine



