
疾病簡介
家族性澱粉樣多發性神經病變(Familial amyloid polyneuropathy, FAP)是指澱粉樣蛋白的基因發生突變,進而造成其製造的蛋白結構異常,沉積於身體各部位,如皮膚、心臟、肝臟、腎臟等組織或器官,組織或器官出現功能異常,導致多重系統的疾病。
目前主要有三種蛋白質的基因突變與家族性澱粉樣多發性病變有關,致病蛋白為運甲狀腺素蛋白(Transthyretin, TTR)、載脂蛋白A-I (Apolipoprotein A-I) 和凝溶膠蛋白(Gelsolin)。
1. 運甲狀腺素蛋白(Transthyretin, TTR)突變:最常見的類型,據研究指出目前在TTR基因上有超過150種突變,其中以Val30Met為廣泛普遍;在臺灣目前病患TTR基因突變型態多為Ala97Ser,而非Val30Met型。
2.載脂蛋白A-1(Apolipoprotein A-1)突變:在APOA1基因上有16種突變與FAP有關,此類型的澱粉樣蛋白多沉積於腎臟、肝臟和胃腸道,導致器官衰竭。
3.凝溶膠蛋白(Gelsolin)突變:主因於GSN基因上第654位置核苷酸有單一突變所致,以顱神經病變及周邊感覺神經病變為主,伴隨皮膚鬆弛及角膜格狀失養的症狀。
臨床症狀
家族性澱粉樣多發性神經病變(Familial amyloid polyneuropathy, FAP)是一種罕見疾病,依不同的疾病類型與種族,其發生率與發病年齡也有所不同。
家族性澱粉樣多發性神經病變(Familial amyloid polyneuropathy, FAP)中較常見的運甲狀腺素蛋白(Transthyretin, TTR)澱粉樣沉積,在臺灣以Ala97Ser基因型別較多,但目前尚未有確認的盛行率與發病年齡之統計。其臨床特徵可包括周圍感覺運動神經病變和自主神經病變,以及非神經病變(心肌病、腎臟病、玻璃體混濁和中樞神經系統澱粉樣變性)。另依類澱粉侵犯的位置分為三型:神經型(Neuropathic form)、心臟型(Cardiac form)和軟腦膜型(Leptomeningeal form)。
運甲狀腺素蛋白澱粉樣神經病變疾病:
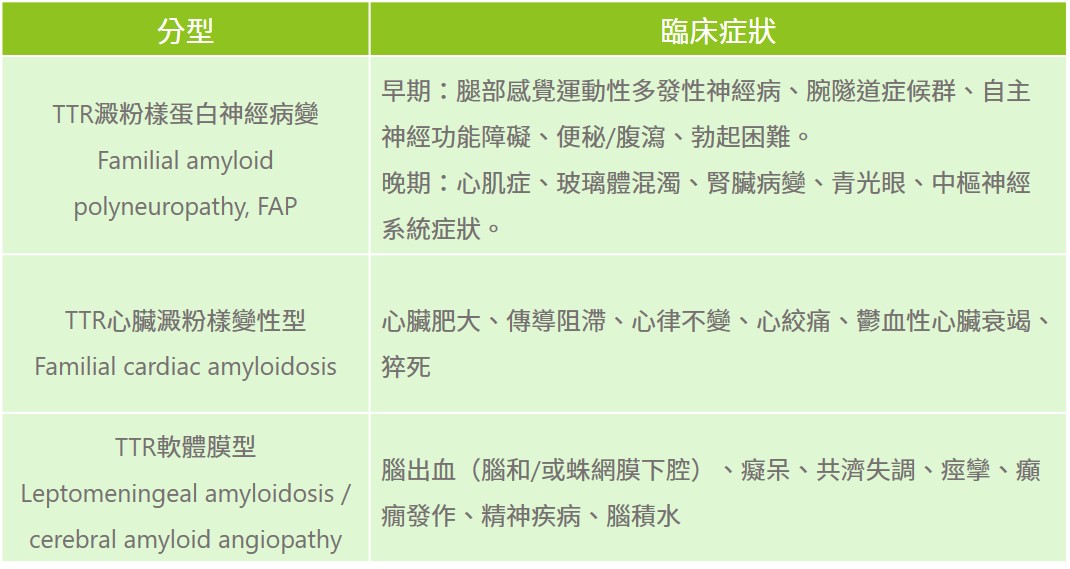
其他遺傳性澱粉樣神經病變疾病:

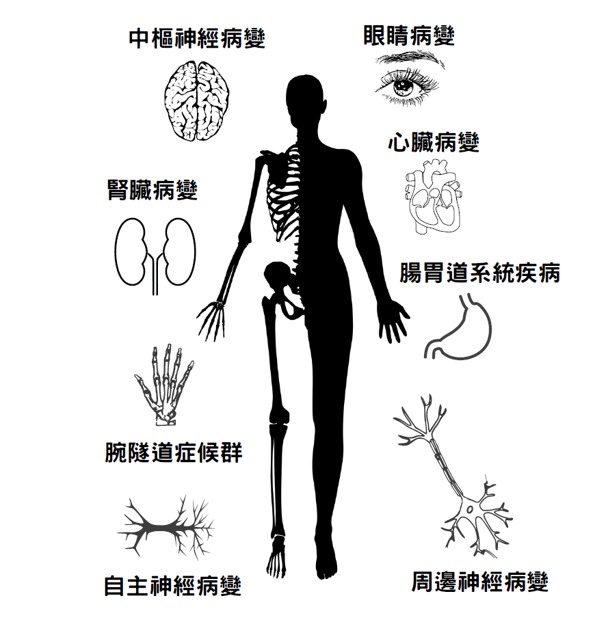
遺傳模式
家族性澱粉樣多發性神經病變(familial amyloid polyneuropathy, FAP)為體染色體顯性之遺傳模式,而夫妻雙方若其一為患者,則下一代有1/2的機率生下患病之胎兒,1/2的機率生下正常之胎兒。
檢驗方式
家族性澱粉樣多發性神經病變(Familial amyloid polyneuropathy, FAP)是一種多重系統的疾病,透過專業醫師進行病理學檢查,確認周邊神經病變、腎臟病變、心臟病變和肝臟病變等症狀,亦可經評估後進行神經電氣生理檢查、心臟超音波、腎臟超音波和血液生化檢驗做為疾病診斷。
針對最常見的TTR基因,也可以透過醫師診斷與評估後利用分子檢測方式進行基因檢測,有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢;確診的患者透過評估可進行手術治療外,目前也已有標靶治療等相關藥物可進行治療。希望透過上述方式,讓患者可以及早診斷與治療。
參考資料
- GeneReviews®
- 財團法人罕見疾病基金會
- 罕見遺傳疾病一點通



