

疾病簡介
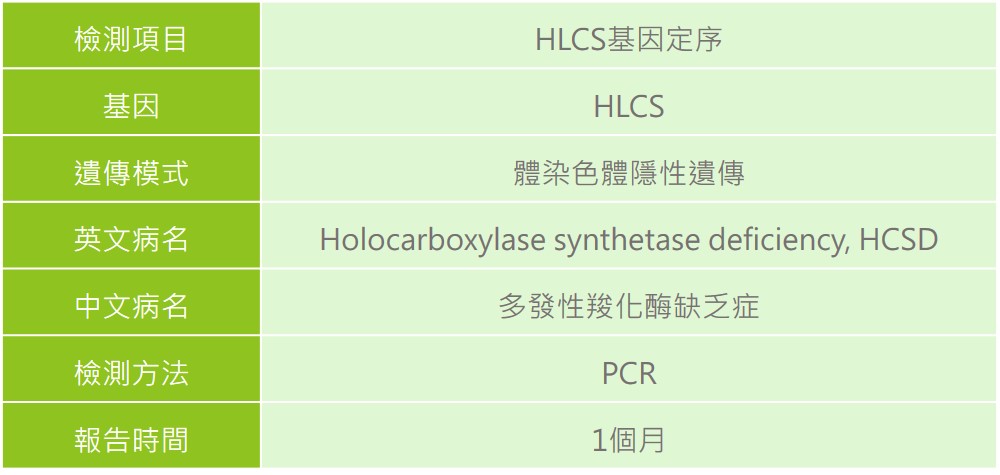
多發性羧化酶缺乏症(Holocarboxylase Synthetase Deficiency, HCSD),致病原因為位於第21號染色體上的HLCS基因突變,使全羧化酶合成酶缺乏,全羧化酶合成酶可以和生物素結合去幫助分解蛋白質、脂質及碳水化合物,一旦HLCS基因產生致病突變,就會降低此酵素結合生物素的能力,造成多種羧化酶無法進行正常功能,因而累積各種異常胺基酸,阻止營養物質的分解,破壞許多細胞功能,而造成症狀表現,通常會在出生後幾個月內即出現症狀。HLCS基因帶因率約為0.7%。
臨床症狀
多發性羧化酶缺乏症患者多數於新生兒或嬰幼兒時期發病,也有部分患者於學齡前才出現症狀;一開始最常發現主要為呼吸方面問題,會有呼吸急促、快速深層呼吸;患者會出現酮酸中毒、有機酸血症及高氨酸血症之症狀,包括嗜睡、張力低下、嘔吐、癲癇、低溫等,嚴重時可能會造成患者昏迷或早夭。在一些生物素治療的早發性或未治療輕微的患者,則可能會出現發展遲緩、掉髮、皮膚病變等症狀。
患者須終生補充生物素以減輕症狀,但若沒有進行治療,此症可能會導致發育遲緩、癲癇發作及昏迷,甚至危及生命安全,另外患者需定期監測血液中生化數值,及生長發育是否有異常的情形。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有 1/4 的機率生下患者。
檢驗方式
多發性羧化酶缺乏症為新生兒篩檢檢測項目之一,測定濾紙血片檢體中C5OH之含量,若濃度高於正常值,需進行複檢,若複檢結果仍為異常時,需至各大醫療中心進行確認診斷。
除了專業醫師的評估外,透過氣相層析質譜儀(GC/MS)檢測尿液中相關代謝有機酸的狀態,及檢測患者血液淋巴球或皮膚纖維母細胞的多發性羧化酶的活性,另外也可以透過分子基因檢測方式進行確認診斷。
針對有臨床特徵之患者、有家族疾病史之家族成員等,建議可與專業醫師進行遺傳諮詢,透過臨床評估與基因檢測的方式,讓多發性羧化酶之患者可以及早診斷治療與醫療協助。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
參考資料
- Pediatr Radiol. 2016;46(3):357-64.
- Mol Genet Metab Rep. 2016;7:40-4.
- 罕見疾病一點通



